AI制药 - AlphaFold Multimer 的 MSA Pairing 源码

目前最新版本是v2.3.1,2023.1.12
-
AlphaFold multimer v1 于 2021 年 7 月发布,同时发表了一篇描述其方法和结果的论文。AlphaFold multimer v1 使用了与 AlphaFold 单体相同的模型结构和训练方法,但增加了一些特征和损失函数来处理多条链。AlphaFold multimer v1 在几个蛋白质复合物的基准测试中取得了最先进的性能。
-
AlphaFold multimer v2 于 2021 年 9 月 21 日发布,作为一个错误修复版本。AlphaFold multimer v2 没有改变模型参数或结构,但修复了松弛阶段的一些问题,并更新了一些第三方库。
-
AlphaFold multimer v3 于 2021 年 12 月 3 日发布,带来了新的模型参数,预计在大型蛋白质复合物上更准确。AlphaFold multimer v3 使用了与 AlphaFold multimer v1 相同的模型结构和训练方法,但使用了不同的数据和超参数。AlphaFold multimer v3 还包括了一些内存优化和可用性改进。
入口函数:run_alphafold.py
调用逻辑:
def predict_structure(fasta_path: str,fasta_name: str,output_dir_base: str,data_pipeline: Union[pipeline.DataPipeline, pipeline_multimer.DataPipeline],model_runners: Dict[str, model.RunModel],amber_relaxer: relax.AmberRelaxation,benchmark: bool,random_seed: int):
其中,data_pipeline,选择pipeline_multimer.DataPipeline:
msa_output_dir = os.path.join(output_dir, 'msas')if not os.path.exists(msa_output_dir):os.makedirs(msa_output_dir)feature_dict = data_pipeline.process(input_fasta_path=fasta_path,msa_output_dir=msa_output_dir)
调用文件:pipeline_multimer.py,其中核心逻辑有三块:
_process_single_chain,单链处理逻辑add_assembly_features,添加特征来区分不同的链pair_and_merge,配对合并MSA
源码及注释如下:
def process(self,input_fasta_path: str,msa_output_dir: str) -> pipeline.FeatureDict:"""Runs alignment tools on the input sequences and creates features."""# 打开输入的fasta文件并读取内容with open(input_fasta_path) as f:input_fasta_str = f.read()# 解析fasta文件中的序列和描述信息input_seqs, input_descs = parsers.parse_fasta(input_fasta_str)# 根据序列和描述信息创建一个链ID的映射表chain_id_map = _make_chain_id_map(sequences=input_seqs,descriptions=input_descs)# 将链ID的映射表保存为json文件chain_id_map_path = os.path.join(msa_output_dir, 'chain_id_map.json')with open(chain_id_map_path, 'w') as f:chain_id_map_dict = {chain_id: dataclasses.asdict(fasta_chain)for chain_id, fasta_chain in chain_id_map.items()}json.dump(chain_id_map_dict, f, indent=4, sort_keys=True)# 初始化一个空字典来存储所有链的特征all_chain_features = {}# 初始化一个空字典来存储已经处理过的序列的特征,避免重复计算sequence_features = {}# 判断输入的序列是否是复合物或单体,即是否只有一种不同的序列is_homomer_or_monomer = len(set(input_seqs)) == 1# 遍历每个链ID和对应的fasta信息for chain_id, fasta_chain in chain_id_map.items():# 如果该链的序列已经在sequence_features中,直接复制其特征到all_chain_features中if fasta_chain.sequence in sequence_features:all_chain_features[chain_id] = copy.deepcopy(sequence_features[fasta_chain.sequence])continue# 否则,调用另一个函数来处理单个链,包括运行比对工具,生成特征等chain_features = self._process_single_chain(chain_id=chain_id,sequence=fasta_chain.sequence,description=fasta_chain.description,msa_output_dir=msa_output_dir,is_homomer_or_monomer=is_homomer_or_monomer)# 将单个链的特征转换为单体特征,即添加一些额外的信息,如链ID等chain_features = convert_monomer_features(chain_features, chain_id=chain_id)# 将单个链的特征添加到all_chain_features中all_chain_features[chain_id] = chain_features# 将单个链的特征添加到sequence_features中,以备后用sequence_features[fasta_chain.sequence] = chain_features# 为所有链的特征添加组装特征,即考虑多个链之间的相互作用等all_chain_features = add_assembly_features(all_chain_features)# 将所有链的特征进行配对和合并,得到一个numpy数组格式的样本np_example = feature_processing.pair_and_merge(all_chain_features=all_chain_features)# 将所有链的特征进行配对和合并,得到一个numpy数组格式的样本# Pad MSA to avoid zero-sized extra_msa.np_example = pad_msa(np_example, 512)# 返回最终的样本return np_example
其中,_process_single_chain的核心逻辑,如下:
- 调用
self._monomer_data_pipeline.process(),生成单链的MSA信息 - 针对于多链,调用
self._all_seq_msa_features。
源码及注释如下:
def _process_single_chain(self,chain_id: str,sequence: str,description: str,msa_output_dir: str,is_homomer_or_monomer: bool) -> pipeline.FeatureDict:"""Runs the monomer pipeline on a single chain."""# 为单个链生成fasta字符串chain_fasta_str = f'>chain_{chain_id}\n{sequence}\n'# 为单个链创建msa输出目录chain_msa_output_dir = os.path.join(msa_output_dir, chain_id)# 如果目录不存在,就创建它if not os.path.exists(chain_msa_output_dir):os.makedirs(chain_msa_output_dir)# 使用临时fasta文件运行单体数据流程with temp_fasta_file(chain_fasta_str) as chain_fasta_path:logging.info('Running monomer pipeline on chain %s: %s',chain_id, description)# 获取单个链的特征字典chain_features = self._monomer_data_pipeline.process(input_fasta_path=chain_fasta_path,msa_output_dir=chain_msa_output_dir)# 如果有两个或更多不同的序列,就构建配对特征# We only construct the pairing features if there are 2 or more unique# sequences.if not is_homomer_or_monomer:all_seq_msa_features = self._all_seq_msa_features(chain_fasta_path,chain_msa_output_dir)# 更新单个链的特征字典chain_features.update(all_seq_msa_features)# 返回单个链的特征字典return chain_features
其中,_all_seq_msa_features的核心逻辑,如下:
- 额外添加MSA的物种信息,即
msa_species_identifiers。
def _all_seq_msa_features(self, input_fasta_path, msa_output_dir):"""Get MSA features for unclustered uniprot, for pairing."""# 为未聚类的uniprot获取msa输出路径out_path = os.path.join(msa_output_dir, 'uniprot_hits.sto')# 运行msa工具,获取sto格式的结果result = pipeline.run_msa_tool(self._uniprot_msa_runner, input_fasta_path, out_path, 'sto',self.use_precomputed_msas)# 解析sto格式的结果,得到msa对象msa = parsers.parse_stockholm(result['sto'])# 截断msa对象,使其序列数不超过最大值msa = msa.truncate(max_seqs=self._max_uniprot_hits)# 从msa对象中提取特征all_seq_features = pipeline.make_msa_features([msa])# 筛选出有效的特征valid_feats = msa_pairing.MSA_FEATURES + ('msa_species_identifiers', # MSA物种标识符)# 为特征添加前缀feats = {f'{k}_all_seq': v for k, v in all_seq_features.items()if k in valid_feats}# 返回特征字典return feats
其中,add_assembly_features的核心逻辑,如下:
- 区分同源二聚体和异源二聚体,使用不同的链名标识。
- 同时,添加不同的特征,用于区分同源和异源,如asym_id、sym_id、entity_id。
def add_assembly_features(all_chain_features: MutableMapping[str, pipeline.FeatureDict],) -> MutableMapping[str, pipeline.FeatureDict]:"""添加特征来区分不同的链。Args:all_chain_features: 一个字典,将链的id映射到每条链的特征字典。Returns:all_chain_features: 一个字典,将形式为`<seq_id>_<sym_id>`的字符串映射到相应的链特征。例如,一个同源二聚体的两条链会有键A_1和A_2。一个异源二聚体的两条链会有键A_1和B_1。"""# 按序列分组链# 创建一个空字典,用来存储序列和实体id的对应关系seq_to_entity_id = {}# 创建一个默认字典,用来按序列分组链的特征grouped_chains = collections.defaultdict(list)# 遍历所有链的特征for chain_id, chain_features in all_chain_features.items():# 获取链的序列seq = str(chain_features['sequence'])# 如果序列不在字典中,就给它分配一个新的实体idif seq not in seq_to_entity_id:seq_to_entity_id[seq] = len(seq_to_entity_id) + 1# 将链的特征添加到对应序列的列表中grouped_chains[seq_to_entity_id[seq]].append(chain_features)# 创建一个新的空字典,用来存储添加了新特征的链new_all_chain_features = {}# 初始化一个链的idchain_id = 1# 遍历按序列分组的链的特征for entity_id, group_chain_features in grouped_chains.items():# 遍历每个序列中的链,给它们分配一个对称idfor sym_id, chain_features in enumerate(group_chain_features, start=1):# 用实体id和对称id构造一个新的键,如A_1或B_2new_all_chain_features[f'{int_id_to_str_id(entity_id)}_{sym_id}'] = chain_features# 获取链的长度seq_length = chain_features['seq_length']# 添加不对称id,对称id和实体id作为特征chain_features['asym_id'] = chain_id * np.ones(seq_length)chain_features['sym_id'] = sym_id * np.ones(seq_length)chain_features['entity_id'] = entity_id * np.ones(seq_length)# 更新链的idchain_id += 1# 返回添加了新特征的链的字典return new_all_chain_features
其中,pair_and_merge的核心逻辑,如下:
process_unmerged_features,合并预处理。create_paired_features,配对特征。deduplicate_unpaired_sequences,移除与配对序列重复的未配对序列merge_chain_features,合并链特征。
def pair_and_merge(all_chain_features: MutableMapping[str, pipeline.FeatureDict]) -> pipeline.FeatureDict:"""对特征进行增强、配对和合并的处理。Args:all_chain_features: 一个可变映射,存储每条链的特征字典。Returns:一个特征字典。"""# 对未合并的特征进行处理process_unmerged_features(all_chain_features)# 将所有链的特征转换为列表np_chains_list = list(all_chain_features.values())# 判断是否需要对MSA序列进行配对, _is_homomer_or_monomer中true是同源,false是异源# pair_msa_sequences表示异源pair_msa_sequences = not _is_homomer_or_monomer(np_chains_list)if pair_msa_sequences: # 异源# 使用msa_pairing模块创建配对的特征np_chains_list = msa_pairing.create_paired_features(chains=np_chains_list)# 去除未配对的重复序列np_chains_list = msa_pairing.deduplicate_unpaired_sequences(np_chains_list)# 裁剪链的长度,限制MSA和模板的数量np_chains_list = crop_chains(np_chains_list,msa_crop_size=MSA_CROP_SIZE,pair_msa_sequences=pair_msa_sequences,max_templates=MAX_TEMPLATES)# 合并链的特征np_example = msa_pairing.merge_chain_features(np_chains_list=np_chains_list, pair_msa_sequences=pair_msa_sequences,max_templates=MAX_TEMPLATES)# 对最终的特征进行处理np_example = process_final(np_example)return np_example
其中,process_unmerged_features的核心逻辑,在chain_features中添加若干特征:
- 包括deletion_matrix、deletion_matrix_all_seq、deletion_mean、all_atom_mask、all_atom_positions、entity_mask
- 与多链相关的assembly_num_chains
源码如下:
def process_unmerged_features(all_chain_features: MutableMapping[str, pipeline.FeatureDict]):"""对合并前的每条链的特征进行后处理。"""num_chains = len(all_chain_features)for chain_features in all_chain_features.values():# 将删除矩阵转换为浮点数。chain_features['deletion_matrix'] = np.asarray(chain_features.pop('deletion_matrix_int'), dtype=np.float32)if 'deletion_matrix_int_all_seq' in chain_features:chain_features['deletion_matrix_all_seq'] = np.asarray(chain_features.pop('deletion_matrix_int_all_seq'), dtype=np.float32)# 计算删除矩阵的均值。chain_features['deletion_mean'] = np.mean(chain_features['deletion_matrix'], axis=0)# 根据aatype添加all_atom_mask和虚拟的all_atom_positions。all_atom_mask = residue_constants.STANDARD_ATOM_MASK[chain_features['aatype']]chain_features['all_atom_mask'] = all_atom_maskchain_features['all_atom_positions'] = np.zeros(list(all_atom_mask.shape) + [3])# 添加assembly_num_chains。chain_features['assembly_num_chains'] = np.asarray(num_chains)# 添加entity_mask。for chain_features in all_chain_features.values():chain_features['entity_mask'] = (chain_features['entity_id'] != 0).astype(np.int32)
其中,merge_chain_features,合并链特征:
- 区分同源体,以及配对和不配对的特征合并。
def merge_chain_features(np_chains_list: List[pipeline.FeatureDict],pair_msa_sequences: bool,max_templates: int) -> pipeline.FeatureDict:"""将多条链的特征合并为单个FeatureDict.参数:np_chains_list: 每条链的FeatureDict的列表.pair_msa_sequences: 是否合并配对的MSA.max_templates: 包含的模板的最大数量.返回:整个复合物的单个FeatureDict."""# 对模板进行填充,使其数量不超过最大值np_chains_list = _pad_templates(np_chains_list, max_templates=max_templates)# 对同源体的密集MSA进行合并np_chains_list = _merge_homomers_dense_msa(np_chains_list)# 不配对的MSA特征将始终被分块对角化;配对的MSA特征将被连接.np_example = _merge_features_from_multiple_chains(np_chains_list, pair_msa_sequences=False)if pair_msa_sequences:# 将配对和不配对的特征连接起来np_example = _concatenate_paired_and_unpaired_features(np_example)# 根据合并后的特征进行修正np_example = _correct_post_merged_feats(np_example=np_example,np_chains_list=np_chains_list,pair_msa_sequences=pair_msa_sequences)return np_example
其中,create_paired_features的核心逻辑,主要步骤:
- 对链进行序列配对,得到配对的行索引:
pair_sequences - 对配对的行进行重新排序:
reorder_paired_rows - 特征填充:
pad_features
def create_paired_features(chains: Iterable[pipeline.FeatureDict]) -> List[pipeline.FeatureDict]:"""返回原始链的特征,其中包含配对的 NUM_SEQ 特征。Args:chains: 每条链的特征字典的列表。Returns:一个特征字典的列表,其中序列特征只包含要配对的行。"""chains = list(chains)chain_keys = chains[0].keys()if len(chains) < 2:return chainselse:updated_chains = []# 对链进行序列配对,得到配对的行索引paired_chains_to_paired_row_indices = pair_sequences(chains)# 对配对的行进行重新排序paired_rows = reorder_paired_rows(paired_chains_to_paired_row_indices)for chain_num, chain in enumerate(chains):# 创建一个新的链特征字典,不包含_all_seq后缀的特征new_chain = {k: v for k, v in chain.items() if '_all_seq' not in k}for feature_name in chain_keys:if feature_name.endswith('_all_seq'):# 对特征进行填充feats_padded = pad_features(chain[feature_name], feature_name)# 只保留配对的行new_chain[feature_name] = feats_padded[paired_rows[:, chain_num]]# 添加num_alignments_all_seq特征new_chain['num_alignments_all_seq'] = np.asarray(len(paired_rows[:, chain_num]))updated_chains.append(new_chain)return updated_chains
其中,pair_sequences的核心逻辑,主要根据物种配对MSA的信息,如下:
- 根据序列相似度匹配MSA行:
_match_rows_by_sequence_similarity
def pair_sequences(examples: List[pipeline.FeatureDict]) -> Dict[int, np.ndarray]:"""返回跨链配对的MSA序列的索引。"""num_examples = len(examples)# 创建一个列表,存储每条链的物种字典all_chain_species_dict = []# 创建一个集合,存储共同的物种common_species = set()for chain_features in examples:# 将链的特征转换为MSA数据框msa_df = _make_msa_df(chain_features)# 根据MSA数据框创建物种字典species_dict = _create_species_dict(msa_df)all_chain_species_dict.append(species_dict)# 将物种字典中的物种添加到共同物种集合中common_species.update(set(species_dict))# 对共同物种进行排序common_species = sorted(common_species)common_species.remove(b'') # 移除目标序列的物种。# 创建一个列表,存储配对的MSA行all_paired_msa_rows = [np.zeros(len(examples), int)]# 创建一个字典,按照出现在多少条链中分组配对的MSA行all_paired_msa_rows_dict = {k: [] for k in range(num_examples)}all_paired_msa_rows_dict[num_examples] = [np.zeros(len(examples), int)]# 遍历共同物种for species in common_species:if not species:continue# 创建一个列表,存储每条链中该物种的MSA数据框this_species_msa_dfs = []# 记录该物种出现在多少条链中species_dfs_present = 0for species_dict in all_chain_species_dict:if species in species_dict:this_species_msa_dfs.append(species_dict[species])species_dfs_present += 1else:this_species_msa_dfs.append(None)# 跳过只出现在一条链中的物种if species_dfs_present <= 1:continue# 跳过MSA数据框过大的物种if np.any(np.array([len(species_df) for species_df inthis_species_msa_dfs ifisinstance(species_df, pd.DataFrame)]) > 600):continue# 根据序列相似度匹配MSA行paired_msa_rows = _match_rows_by_sequence_similarity(this_species_msa_dfs)# 将匹配的MSA行添加到列表和字典中all_paired_msa_rows.extend(paired_msa_rows)all_paired_msa_rows_dict[species_dfs_present].extend(paired_msa_rows)# 将字典中的值转换为数组all_paired_msa_rows_dict = {num_examples: np.array(paired_msa_rows) fornum_examples, paired_msa_rows in all_paired_msa_rows_dict.items()}return all_paired_msa_rows_dict
其中,_match_rows_by_sequence_similarity的核心逻辑:
def _match_rows_by_sequence_similarity(this_species_msa_dfs: List[pd.DataFrame]) -> List[List[int]]:"""根据序列相似度找出跨链的MSA序列配对。首先,将每条链的MSA序列按照它们与各自目标序列的相似度进行排序。然后,从最相似的序列开始进行配对。Args:this_species_msa_dfs: 一个列表,包含了特定物种的MSA特征的数据框。Returns:一个列表的列表,每个列表包含M个索引,对应于配对的MSA行,其中M是链的数量。"""all_paired_msa_rows = []# 获取每个数据框中的序列数量num_seqs = [len(species_df) for species_df in this_species_msa_dfsif species_df is not None]# 取最小的序列数量take_num_seqs = np.min(num_seqs)# 定义一个函数,按照相似度对数据框进行排序sort_by_similarity = (lambda x: x.sort_values('msa_similarity', axis=0, ascending=False))for species_df in this_species_msa_dfs:if species_df is not None:# 对该物种的数据框进行排序species_df_sorted = sort_by_similarity(species_df)# 获取前take_num_seqs个MSA行的索引msa_rows = species_df_sorted.msa_row.iloc[:take_num_seqs].valueselse:# 如果该物种不存在,则取最后一行(填充行)的索引msa_rows = [-1] * take_num_seqs all_paired_msa_rows.append(msa_rows)# 将所有链的MSA行索引转置all_paired_msa_rows = list(np.array(all_paired_msa_rows).transpose())return all_paired_msa_rows
其中,deduplicate_unpaired_sequences的核心逻辑:
def deduplicate_unpaired_sequences(np_chains: List[pipeline.FeatureDict]) -> List[pipeline.FeatureDict]:"""移除与配对序列重复的未配对序列。"""# 获取特征的名称feature_names = np_chains[0].keys()# 获取MSA相关的特征msa_features = MSA_FEATURESfor chain in np_chains:# 将msa_all_seq特征转换为元组,方便哈希sequence_set = set(tuple(s) for s in chain['msa_all_seq'])keep_rows = []# 遍历未配对的MSA序列,移除任何与已配对序列相同的行for row_num, seq in enumerate(chain['msa']):if tuple(seq) not in sequence_set:keep_rows.append(row_num)# 更新MSA相关的特征,只保留需要的行for feature_name in feature_names:if feature_name in msa_features:chain[feature_name] = chain[feature_name][keep_rows]# 更新num_alignments特征chain['num_alignments'] = np.array(chain['msa'].shape[0], dtype=np.int32)return np_chains
其中,reorder_paired_rows的核心逻辑:
- 创建一个包含跨链配对MSA行的索引列表.
- 对配对链的数量进行降序遍历
def reorder_paired_rows(all_paired_msa_rows_dict: Dict[int, np.ndarray]) -> np.ndarray:"""创建一个包含跨链配对MSA行的索引列表.参数:all_paired_msa_rows_dict: 一个映射,从配对链的数量到配对索引.返回:一个列表的列表,每个列表包含跨链配对MSA行的索引.配对索引列表按以下顺序排序:1) 配对比对中的链的数量,即,所有链的配对将排在前面.2) e值"""# 初始化一个空列表all_paired_msa_rows = []# 对配对链的数量进行降序遍历for num_pairings in sorted(all_paired_msa_rows_dict, reverse=True):# 获取当前数量的配对索引paired_rows = all_paired_msa_rows_dict[num_pairings]# 计算每个配对索引的乘积的绝对值paired_rows_product = abs(np.array([np.prod(rows) for rows in paired_rows]))# 按照乘积的大小进行升序排序paired_rows_sort_index = np.argsort(paired_rows_product)# 将排序后的配对索引添加到列表中all_paired_msa_rows.extend(paired_rows[paired_rows_sort_index])# 将列表转换为数组并返回return np.array(all_paired_msa_rows)
相关文章:
AI制药 - AlphaFold Multimer 的 MSA Pairing 源码
目前最新版本是v2.3.1,2023.1.12 AlphaFold multimer v1 于 2021 年 7 月发布,同时发表了一篇描述其方法和结果的论文。AlphaFold multimer v1 使用了与 AlphaFold 单体相同的模型结构和训练方法,但增加了一些特征和损失函数来处理多条链。Al…...

TitanIDE:云原生开发到底强在哪里?
原文作者:行云创新技术总监 邓冰寒 引言 是一种新的软件开发方法,旨在构建更可靠、高效、弹性、安全和可扩展的应用程序。与传统的应用程序开发方式不同,云原生是将开发环境完全搬到云端,构建一站式的云原生开发环境。云原生的开…...

单片机常用完整性校验算法
一、前言 单片机在开发过程中经常会遇到大文件传输,或者大量数据传输,在一些工业环境下,数据传输并不是很稳定,如何检验数据的完整性就是个问题,这里简单介绍一下单片机常用的几种数据完整性校验方法。 二、CheckSum校…...
Anaconda 的安装配置及依赖项的内外网配置
在分享anaconda 的安装配置及使用前,我们必须先明白anaconda是什么;Anaconda是一个开源的Python发行版本。两者区别在于前者是一门编程语言,后者相当于编程语言中的工具包。 由于python自身缺少numpy、matplotlib、scipy、scikit-learn等一系…...
p84 CTF夺旗-PHP弱类型异或取反序列化RCE
数据来源 文章参考 本课重点: 案例1:PHP-相关总结知识点-后期复现案例2:PHP-弱类型对比绕过测试-常考点案例3:PHP-正则preg_match绕过-常考点案例4:PHP-命令执行RCE变异绕过-常考点案例5:PHP-反序列化考题…...

2022财报逆转,有赞穿透迷雾实现突破
2022年,商家经营面临困难。但在一些第三方服务商的帮助下,也有商家取得了逆势增长。 2023年3月23日,有赞发布2022年业绩报告,它帮助许多商家稳住了一整年的经营。2022年,有赞门店SaaS业务的GMV达到425亿元,…...

蓝桥杯 - 求组合数【C(a,b)】+ 卡特兰数
文章目录💬前言885. 求组合数 I C(m,n) 【dp】886 求组合数 II 【数据大小10万级别】 【费马小定理快速幂逆元】887. 求组合数 III 【le18级别】 【卢卡斯定理 逆元 快速幂 】888.求组合数 IV 【没有%p -- 高精度算出准确结果】 【分解质因数 高精度乘法 --只用一…...

膳食真菌在癌症免疫治疗中的作用: 从肠道微生物群的角度
谷禾健康 癌症是一种恶性肿瘤,它可以发生在人体的任何部位,包括肺、乳房、结肠、胃、肝、宫颈等。根据世界卫生组织的数据,全球每年有超过1800万人被诊断出患有癌症,其中约有1000万人死于癌症。癌症已成为全球范围内的主要健康问题…...

怎么将模糊的照片变清晰
怎么将模糊的照片变清晰?珍贵的照片每个人都会有,而遇到珍贵的照片变模糊了,相信会让人很苦恼的。那么有没有办法可以解决呢?答案是有的,我们可以用工具让模糊的照片变得清晰。下面就来分享一些让模糊的照片变清晰的方法,有兴趣…...
【软件测试】基础知识第一篇
文章目录一. 什么是软件测试二. 测试和调试的区别三. 什么是测试用例四. 软件的生命周期五. 软件测试的生命周期一. 什么是软件测试 软件测试就是验证软件产品特性是否满足用户的需求。 那需求又是什么呢?在多数软件公司,会有两种需求,一种…...

【百面成神】java web基础7问,你能坚持到第几问
前 言 🍉 作者简介:半旧518,长跑型选手,立志坚持写10年博客,专注于java后端 ☕专栏简介:纯手打总结面试题,自用备用 🌰 文章简介:java web最基础、重要的8道面试题 文章目…...
Centos7安装、各种环境配置和常见bug解决方案,保姆级教程(更新中)
文章目录前言一、Centos7安装二、各种环境配置与安装2.1 安装net-tools(建议)2.2 配置静态网络(建议)2.1 修改Centos7的时间(建议)2.2 Centos7系统编码问题2.3 vim安装(建议)2.4 解决…...

【C++进阶】智能指针
文章目录为什么需要智能指针?内存泄漏什么是内存泄漏,内存泄漏的危害内存泄漏分类(了解)如何避免内存泄漏智能指针的使用及原理smart_ptrauto_ptrunique_ptrshared_ptr线程安全的解决循环引用weak_ptr删除器为什么需要智能指针&am…...

软件测试面试题 —— 整理与解析(3)
😏作者简介:博主是一位测试管理者,同时也是一名对外企业兼职讲师。 📡主页地址:🌎【Austin_zhai】🌏 🙆目的与景愿:旨在于能帮助更多的测试行业人员提升软硬技能…...

springboot常用的20个注解
Spring Boot方式的项目开发已经逐步成为Java应用开发领域的主流框架,它不仅可以方便地创建生产级的Spring应用程序,还能轻松地通过一些注解配置与目前比较火热的微服务框架SpringCloud集成, 而Spring Boot 之所以能够轻松地实现应的创建及与…...

USB组合设备——带鼠标功能的键盘
文章目录带鼠标功能的键盘一个接口实现报告描述符示例多个接口实现复合设备和组合设备配置描述符集合的实现报告的返回附 STM32 枚举日志复合设备:Compound Device 内嵌 Hub 和多个 Function,每个 Function 都相当于一个独立的 USB 外设,有自…...
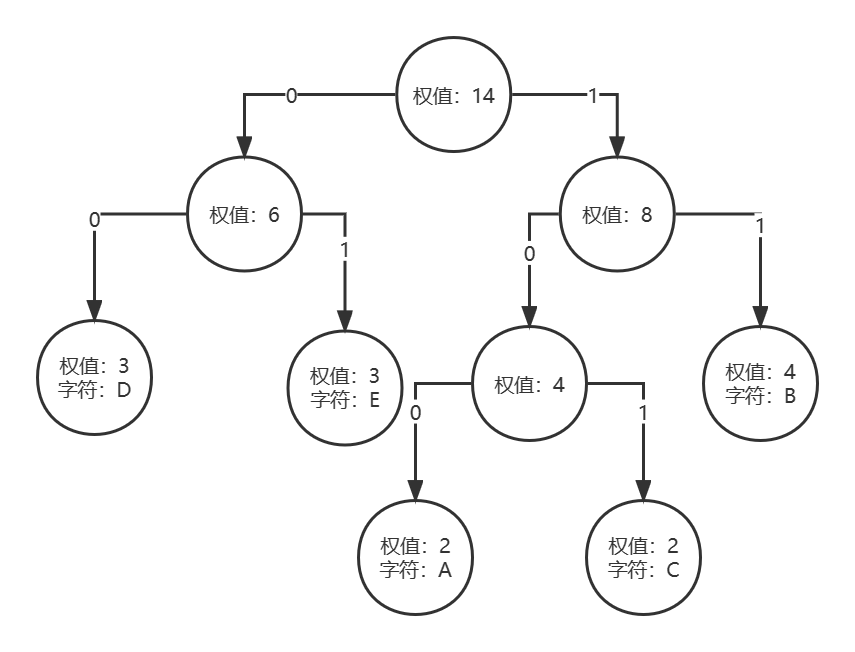
数据结构与算法基础-学习-18-哈夫曼编码
一、个人理解在远程通讯中,需要把字符转成二进制的字符串进行传输,例如我们需要传输ABCD,我们可以用定长的字符串进行表示,例如:A:00B:01C:02D:03这样可能就造成空间的浪费,我们多存储了一个0号位。那用变长呢…...

ZMC408CE | 实现“8通道独立PSO”应用场景
一、ZMC408SCAN产品亮点 1.高性能处理器,提升运算速度、响应时间和扫描周期等; 2.一维/二维/三维、多通道视觉飞拍,高速高精; 3.位置同步输出PSO,连续轨迹加工中对精密点胶胶量控制和激光能量控制等; 4…...

QuickJS中JS_SetClassProto方法把JavaScript对象指定为某个类的原型对象
在 QuickJS 中,JS_SetClassProto 方法用于设置一个类的原型对象。这个方法的作用是将一个 JavaScript 对象指定为该类的原型对象,从而定义该类的属性和方法。 具体来说,JS_SetClassProto 方法的第一个参数是指向 QuickJS 引擎执行上下文的指…...

泰克信号发生器特点
泰克信号发生器是一种用于产生各种类型的电子信号的仪器,可以广泛应用于电子、通信、自动化、医疗等领域。泰克信号发生器具有以下特点:多种信号类型:泰克信号发生器可以产生多种类型的电子信号,包括正弦波、方波、三角波、脉冲等…...

线程同步:确保多线程程序的安全与高效!
全文目录: 开篇语前序前言第一部分:线程同步的概念与问题1.1 线程同步的概念1.2 线程同步的问题1.3 线程同步的解决方案 第二部分:synchronized关键字的使用2.1 使用 synchronized修饰方法2.2 使用 synchronized修饰代码块 第三部分ÿ…...

解锁数据库简洁之道:FastAPI与SQLModel实战指南
在构建现代Web应用程序时,与数据库的交互无疑是核心环节。虽然传统的数据库操作方式(如直接编写SQL语句与psycopg2交互)赋予了我们精细的控制权,但在面对日益复杂的业务逻辑和快速迭代的需求时,这种方式的开发效率和可…...
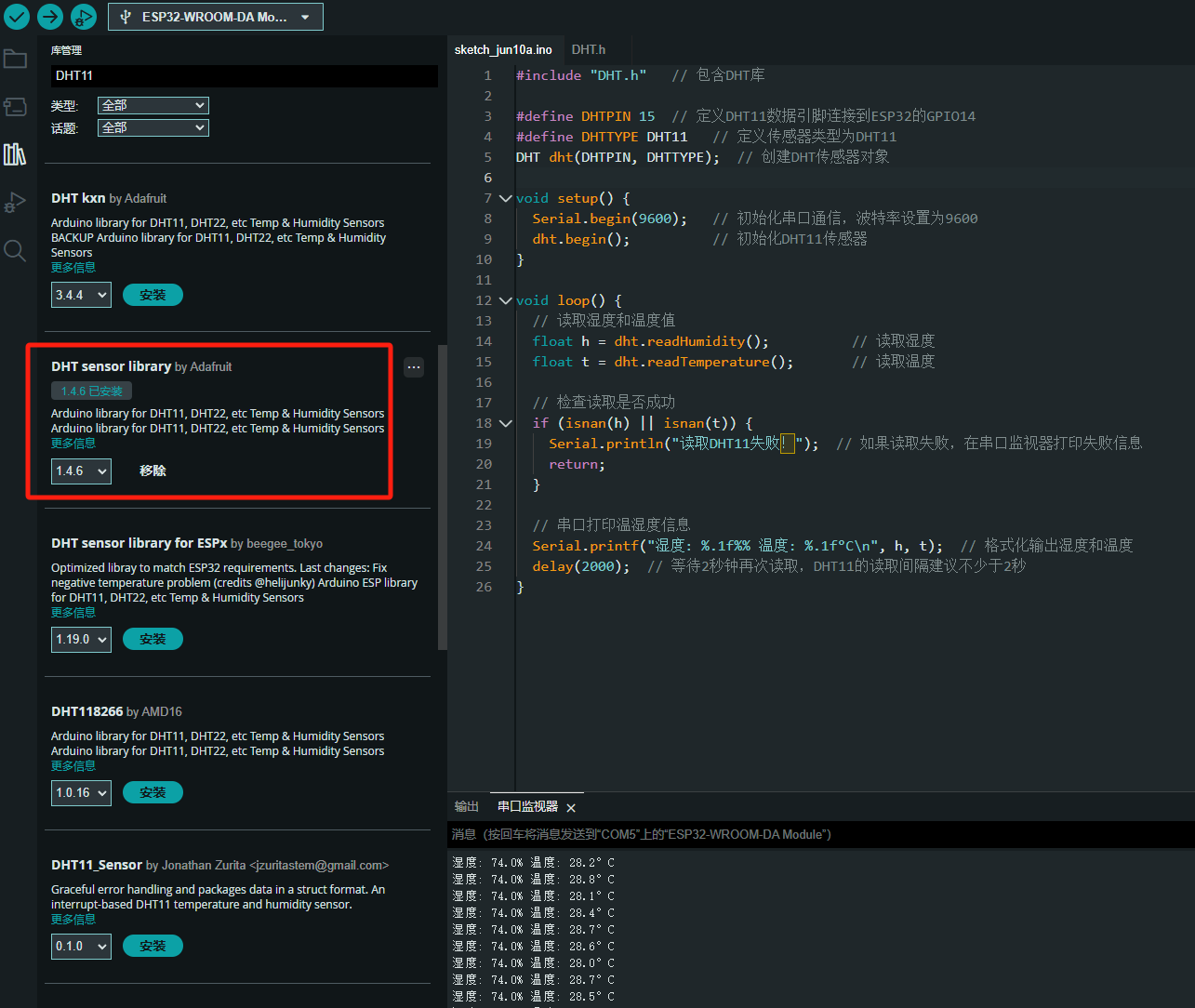
ESP32读取DHT11温湿度数据
芯片:ESP32 环境:Arduino 一、安装DHT11传感器库 红框的库,别安装错了 二、代码 注意,DATA口要连接在D15上 #include "DHT.h" // 包含DHT库#define DHTPIN 15 // 定义DHT11数据引脚连接到ESP32的GPIO15 #define D…...
-----深度优先搜索(DFS)实现)
c++ 面试题(1)-----深度优先搜索(DFS)实现
操作系统:ubuntu22.04 IDE:Visual Studio Code 编程语言:C11 题目描述 地上有一个 m 行 n 列的方格,从坐标 [0,0] 起始。一个机器人可以从某一格移动到上下左右四个格子,但不能进入行坐标和列坐标的数位之和大于 k 的格子。 例…...
Mac软件卸载指南,简单易懂!
刚和Adobe分手,它却总在Library里给你写"回忆录"?卸载的Final Cut Pro像电子幽灵般阴魂不散?总是会有残留文件,别慌!这份Mac软件卸载指南,将用最硬核的方式教你"数字分手术"࿰…...

Unit 1 深度强化学习简介
Deep RL Course ——Unit 1 Introduction 从理论和实践层面深入学习深度强化学习。学会使用知名的深度强化学习库,例如 Stable Baselines3、RL Baselines3 Zoo、Sample Factory 和 CleanRL。在独特的环境中训练智能体,比如 SnowballFight、Huggy the Do…...

【生成模型】视频生成论文调研
工作清单 上游应用方向:控制、速度、时长、高动态、多主体驱动 类型工作基础模型WAN / WAN-VACE / HunyuanVideo控制条件轨迹控制ATI~镜头控制ReCamMaster~多主体驱动Phantom~音频驱动Let Them Talk: Audio-Driven Multi-Person Conversational Video Generation速…...
LINUX 69 FTP 客服管理系统 man 5 /etc/vsftpd/vsftpd.conf
FTP 客服管理系统 实现kefu123登录,不允许匿名访问,kefu只能访问/data/kefu目录,不能查看其他目录 创建账号密码 useradd kefu echo 123|passwd -stdin kefu [rootcode caozx26420]# echo 123|passwd --stdin kefu 更改用户 kefu 的密码…...

【堆垛策略】设计方法
堆垛策略的设计是积木堆叠系统的核心,直接影响堆叠的稳定性、效率和容错能力。以下是分层次的堆垛策略设计方法,涵盖基础规则、优化算法和容错机制: 1. 基础堆垛规则 (1) 物理稳定性优先 重心原则: 大尺寸/重量积木在下…...
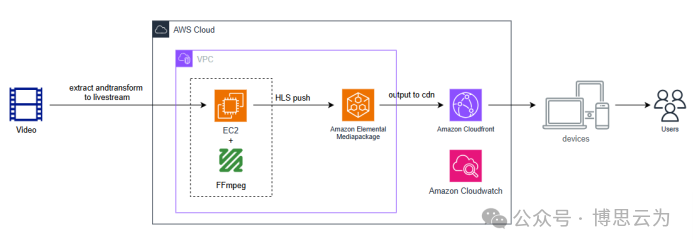
客户案例 | 短视频点播企业海外视频加速与成本优化:MediaPackage+Cloudfront 技术重构实践
01技术背景与业务挑战 某短视频点播企业深耕国内用户市场,但其后台应用系统部署于东南亚印尼 IDC 机房。 随着业务规模扩大,传统架构已较难满足当前企业发展的需求,企业面临着三重挑战: ① 业务:国内用户访问海外服…...