PACNet CellNet(代码开源)|bulk数据作细胞分类,评估细胞命运性能的一大利器
文章目录
- 1.前言
- 2.CellNet
- 2.1CellNet简介
- 2.2CellNet结果
- 3.PACNet
- 3.1安装R包与加载R包
- 3.2加载数据
- 3.3开始训练和分类
- 3.4可视化分类过程
- 3.5可视化分类结果
- 4.细胞命运分类和免疫浸润比较
1.前言
今天冲浪看到一个细胞分类性能评估的R包——PACNet,它与转录组分析方法、计算预处理方法和预处理方法产生的基因可用性无关,因此可以对细胞命运工程方案的性能进行交叉研究比较,这个是新包。

其次还有孪生子弟旧R包,已经停止更新了:CellNet

先讲一下CellNet,因为新包的参考数据集也是共用的,但使用的话我们还是用PACNet哈
2.CellNet
2.1CellNet简介
CellNet是一个基于网络生物学的计算平台,用于评估细胞工程的保真度,并生成用于改进细胞衍生的假设。CellNet基于细胞类型特异性基因调控网络(GRN)的重建,有16种小鼠和16种人类细胞和组织类型的公开RNA-Seq数据进行重建。
简单过一下CellNet,目前,有两种方法可以运行CellNet获取RNA-Seq数据。作者提供了云平台和本地运行两种方式,因为亚马逊云要氪金,本着白嫖的意思就本地跑一跑了
- Cutadapt
- Salmon
- GNU Parallel
CellNet的核心是随机森林分类器。这是对细胞命运实验结果进行分类的算法。要使用 CellNet 分析自己的表达数据,需要一个经过训练的 CellNet 分类器对象,我们将其称为 cnProc(CellNet 处理器)。
第一步需要先构建cnProc对象,可以自己去构建,相关代码:构建cnProc,优势就是可以增加自己需要研究细胞类型,或者研究人鼠外其他物种。
可以使用作者整理好了的rdata,在github中下载即可:

第二第三步就是RNA数据(要SRA文件)和索引了
作者也提供了:

2.2CellNet结果
- 分类热图:在训练数据(行)中显示每个样本(列)对每个细胞和组织类型的分类分数:
pdf(file='hmclass_example.pdf', width=7, height=5)
cn_HmClass(cnRes)
dev.off()
这里可以看到iPSC在胚胎干细胞中分数更高,其次是Day0的几个在成纤维细胞中分数更高。

- Gene Regulatory Network 状态栏图:一种更灵敏的测量,用于测量特定细胞类型的 GRN 在实验数据中建立的程度
fname<-'grnstats_esc_example.pdf'
bOrder<-c("fibroblast_train", unique(as.vector(stQuery$description1)), "esc_train")
cn_barplot_grnSing(cnRes,cnProc,"esc", c("fibroblast","esc"), bOrder, sidCol="sra_id")
ggplot2::ggsave(fname, width=5.5, height=5)
dev.off()
这里关于基因调控网络状态的,如果说热图是一个计算分数绝对值的匹配,那这里就是对调控网络状态,一个动态的匹配

- Network Influence Score Box and Whisker Plot:可以更好地调节的转录因子的建议,按其潜在影响进行排序
rownames(stQuery)<-as.vector(stQuery$sra_id)
tfScores<-cn_nis_all(cnRes, cnProc, "esc") fname<-'nis_esc_example_Day0.pdf'
plot_nis(tfScores, "esc", stQuery, "Day0", dLevel="description1", limitTo=0)
ggplot2::ggsave(fname, width=4, height=12)
dev.off()
这个就调控影响分数的排序

3.PACNet
流程和输入文件是与CellNet一样的,直接跳过开始demo
3.1安装R包与加载R包
install.packages("devtools")
library(devtools)
install_github("pcahan1/CellNet", ref="master")
install_github("pcahan1/cancerCellNet@v0.1.1", ref="master")
source("pacnet_utils.R")library(CellNet)
library(cancerCellNet)
library(plyr)
library(ggplot2)
library(RColorBrewer)
library(pheatmap)
library(plotly)
library(igraph)
3.2加载数据
这里就需要表达矩阵和元数据列表。
表达矩阵应将基因符号作为行名,将样本名称作为列名。示例元数据表应将示例名称作为行名,将示例要素作为列名。表达式矩阵的列名必须与元数据表的行名匹配。为了使分类器训练可靠,每种训练类型至少应有 60 个独立rep。
元数据列表的title格式:

加载数据
expTrain <- utils_loadObject("Hs_expTrain_Jun-20-2017.rda")
stTrain <- utils_loadObject("Hs_stTrain_Jun-20-2017.rda")
加载工程参考数据和查询数据
liverRefExpDat <- utils_loadObject("liver_engineeredRef_normalized_expDat_all.rda")
liverRefSampTab <- utils_loadObject("liver_engineeredRef_sampTab_all.rda")
queryExpDat <- read.csv("example_data/example_counts_matrix.csv", row.names=1)
querySampTab <- read.csv("example_data/example_sample_metadata_table.csv")
rownames(querySampTab) <- querySampTab$sample_name
study_name <- "liver_example"
识别交叉基因:
iGenes <- intersect(rownames(expTrain), rownames(liverRefExpDat))
iGenes <- intersect(iGenes, rownames(queryExpDat))
# Subset training expression matrix based on iGenes
expTrain <- expTrain[iGenes,]
3.3开始训练和分类
将数据拆分为训练集和验证集:
set.seed(99) # Setting a seed for the random number generator allows us to reproduce the same split in the future
stList <- splitCommon_proportion(sampTab = stTrain, proportion = 0.66, dLevel = "description1") # Use 2/3 of training data for training and 1/3 for validation
stTrainSubset <- stList$trainingSet
expTrainSubset <- expTrain[,rownames(stTrainSubset)]#See number of samples of each unique type in description1 in training subset
table(stTrainSubset$description1)stValSubset <- stList$validationSet
expValSubset <- expTrain[,rownames(stValSubset)]
#See number of samples of each unique type in description1 in validation subset
table(stValSubset$description1)
训练随机森林分类器,需要 3-10 分钟,具体取决于内存可用性:
system.time(my_classifier <- broadClass_train(stTrain = stTrainSubset, expTrain = expTrainSubset, colName_cat = "description1", colName_samp = "sra_id", nRand = 70, nTopGenes = 100, nTopGenePairs = 100, nTrees = 2000, stratify=TRUE, sampsize=25, # Must be less than the smallest number in table(stTrainSubset$description1)quickPairs=TRUE)) # Increasing the number of top genes and top gene pairs increases the resolution of the classifier but increases the computing time
save(my_classifier, file="example_outputs/cellnet_classifier_100topGenes_100genePairs.rda")
3.4可视化分类过程
- 分类热图:
stValSubsetOrdered <- stValSubset[order(stValSubset$description1), ] #order samples by classification name
expValSubset <- expValSubset[,rownames(stValSubsetOrdered)]
cnProc <- my_classifier$cnProc #select the cnProc from the earlier class trainingclassMatrix <- broadClass_predict(cnProc, expValSubset, nrand = 60)
stValRand <- addRandToSampTab(classMatrix, stValSubsetOrdered, desc="description1", id="sra_id")grps <- as.vector(stValRand$description1)
names(grps)<-rownames(stValRand)# Create validation heatmap
png(file="classification_validation_hm.png", height=6, width=10, units="in", res=300)
ccn_hmClass(classMatrix, grps=grps, fontsize_row=10)
dev.off()

- 验证精度-召回率曲线:
assessmentDat <- ccn_classAssess(classMatrix, stValRand, classLevels="description1", dLevelSID="sra_id")
png(file="example_outputs/classifier_assessment_PR.png", height=8, width=10, units="in", res=300)
plot_class_PRs(assessmentDat)
dev.off()

- 基因对验证:
genePairs <- cnProc$xpairs
# Get gene to gene comparison of each gene pair in the expression table
expTransform <- query_transform(expTrainSubset, genePairs)
avgGenePair_train <- avgGeneCat(expDat = expTransform, sampTab = stTrainSubset, dLevel = "description1", sampID = "sra_id")genePairs_val <- query_transform(expValSubset, genePairs)
geneCompareMatrix <- makeGeneCompareTab(queryExpTab = genePairs_val,avgGeneTab = avgGenePair_train, geneSamples = genePairs)
val_grps <- stValSubset[,"description1"]
val_grps <- c(val_grps, colnames(avgGenePair_train))
names(val_grps) <- c(rownames(stValSubset), colnames(avgGenePair_train))png(file="example_outputs/validation_gene-pair_comparison.png", width=10, height=80, units="in", res=300)
plotGeneComparison(geneCompareMatrix, grps = val_grps, fontsize_row = 6)
dev.off()
该图较大,主要就是基因对的分数热图,这个就是xpairs图

创建并保存xpairs_list对象,用于 grn 重建和训练规范化参数:
xpairs_list <- vector("list", 14)
for (pair in rownames(avgGenePair_train)) {for (j in 1:ncol(avgGenePair_train)) {if (avgGenePair_train[pair,j] >= 0.5) {if (is.null(xpairs_list[[j]])) {xpairs_list[[j]] <- c(pair)} else { xpairs_list[[j]] <- c(xpairs_list[[j]], pair)}} }
}
xpair_names <- colnames(avgGenePair_train)
xpair_names <- sub(pattern="_Avg", replacement="", x=xpair_names)
names(xpairs_list) <- xpair_namesfor (type in names(xpairs_list)) {names(xpairs_list[[type]]) <- xpairs_list[[type]]
}
save(xpairs_list, file="example_outputs/Hs_xpairs_list.rda")
3.5可视化分类结果
对训练集样本进行分类
classMatrixLiverRef <- broadClass_predict(cnProc = cnProc, expDat = liverRefExpDat, nrand = 10)
grp_names1 <- c(as.character(liverRefSampTab$description1), rep("random", 10))
names(grp_names1) <- c(as.character(rownames(liverRefSampTab)), paste0("rand_", c(1:10)))# Re-order classMatrixQuery to match order of rows in querySampTab
classMatrixLiverRef <- classMatrixLiverRef[,names(grp_names1)]png(file="example_outputs/heatmapLiverRef.png", height=12, width=9, units="in", res=300)
heatmapRef(classMatrixLiverRef, liverRefSampTab) # This function can be found in pacnet_utils.R
dev.off()# Alternatively, for an interactive plotly version:
heatmapPlotlyRef(classMatrixLiverRef, liverRefSampTab)

对测试集进行分类:
queryExpDat <- log(1+queryExpDat)
classMatrixQuery <- broadClass_predict(cnProc = cnProc, expDat = queryExpDat, nrand = 3)
grp_names <- c(as.character(querySampTab$description1), rep("random", 3))
names(grp_names) <- c(as.character(rownames(querySampTab)), paste0("rand_", c(1:3)))# Re-order classMatrixQuery to match order of rows in querySampTab
classMatrixQuery <- classMatrixQuery[,names(grp_names)]save(classMatrixQuery, file="example_outputs/example_classificationMatrix.rda")png(file="example_outputs/query_classification_heatmap.png", height=4, width=8, units="in", res=300)
# This function can be found in pacnet_utils.R
acn_queryClassHm(classMatrixQuery, main = paste0("Classification Heatmap, ", study_name), grps = grp_names, fontsize_row=10, fontsize_col = 10, isBig = FALSE)
dev.off()

计算调控因子得分
## 准备用于网络影响得分计算的 GRN 和表达式数据
## 基于交叉基因的子集和对象:grnAll,trainNormParam
grnAll <- utils_loadObject("liver_grnAll.rda")
trainNormParam <- utils_loadObject("liver_trainNormParam.rda")# These two functions can be found in pacnet_utils.R
grnAll <- subsetGRNall(grnAll, iGenes)
trainNormParam <- subsetTrainNormParam(trainNormParam, grnAll, iGenes)queryExpDat_ranked <- logRank(queryExpDat, base = 0)
queryExpDat_ranked <- as.data.frame(queryExpDat_ranked)## 计算转录调节因子的计算网络影响评分 (NIS)
network_cell_type <- "liver"
target_cell_type <- "liver"
system.time(TF_scores <- pacnet_nis(expDat = queryExpDat_ranked, stQuery=querySampTab, iGenes=iGenes,grnAll = grnAll, trainNorm = trainNormParam,subnet = network_cell_type, ctt=target_cell_type,colname_sid="sample_name", relaWeight=0))save(TF_scores, file="example_outputs/my_study_TF_scores.rda")## 选择得分最高的 25 个 TF 进行绘图:
TFsums <- rowSums(abs(TF_scores))
ordered_TFsums <- TFsums[order(TFsums, decreasing = TRUE)]
if(length(TFsums) > 25) {top_display_TFs <- names(ordered_TFsums)[1:25]
} else {top_display_TFs <- names(ordered_TFsums)
}
TF_scores <- TF_scores[top_display_TFs,]## 绘制 TF 分数:
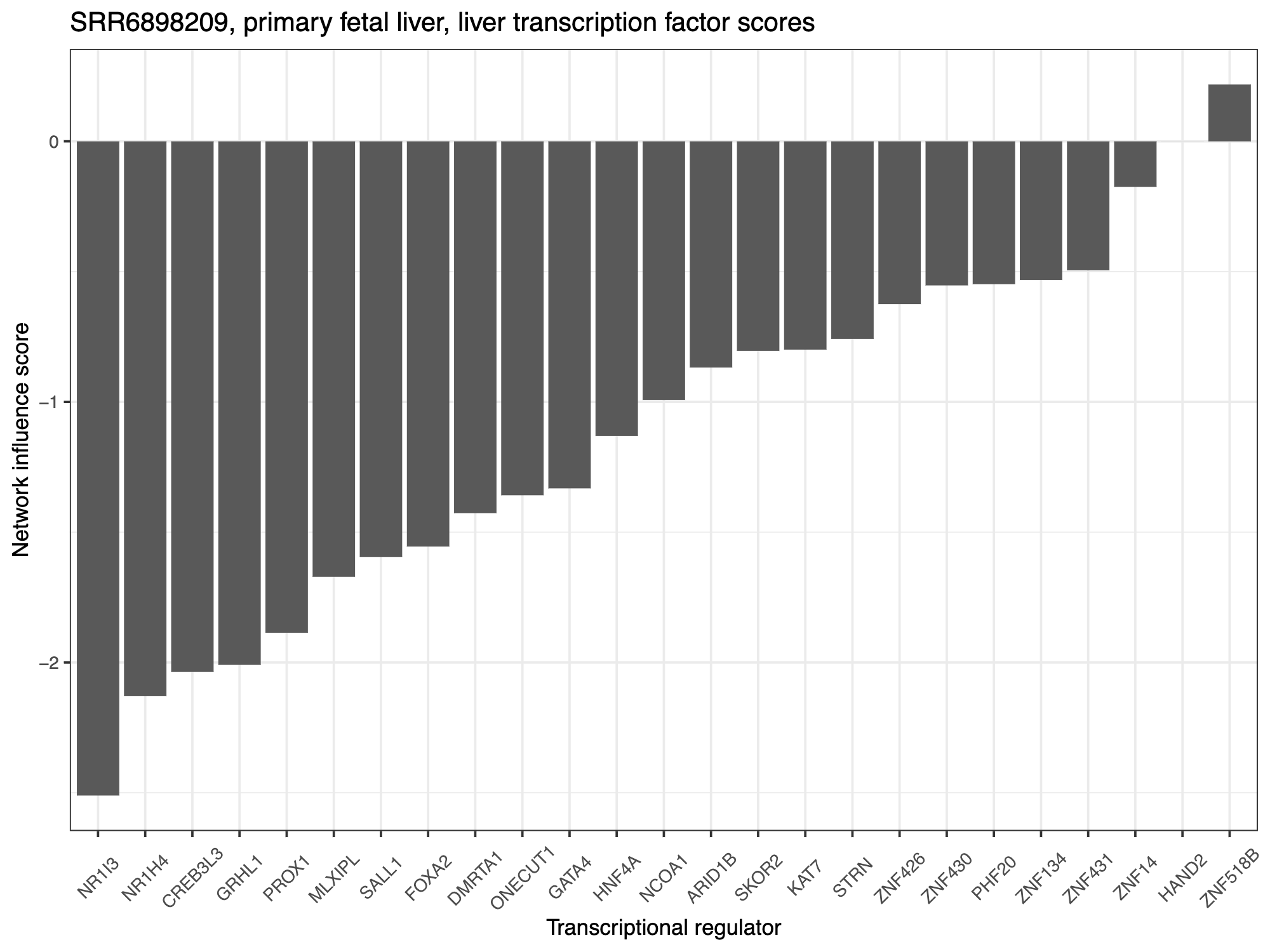
sample_names <- rownames(querySampTab)pdf(file="example_outputs/my_study_TF_scores_my_cell_type.pdf", height=6, width=8)
for(sample in sample_names) {descript <- querySampTab$description1[which(rownames(querySampTab) == sample)]plot_df <- data.frame("TFs" = rownames(TF_scores),"Scores" = as.vector(TF_scores[,sample]))sample_TFplot <- ggplot(plot_df, aes(x = reorder(TFs,Scores,mean) , y = Scores)) + geom_bar(stat="identity") + #aes(fill = medVal)) +theme_bw() + ggtitle(paste0(sample, ", ", descript, ", ", target_cell_type, " transcription factor scores")) +ylab("Network influence score") + xlab("Transcriptional regulator") + theme(legend.position = "none", axis.text = element_text(size = 8)) +theme(text = element_text(size=10), legend.position="none",axis.text.x = element_text(angle = 45, vjust=0.5))print(sample_TFplot)
}
dev.off()
阴性 TF 评分表明给定的 TF 应该上调以获得与靶细胞类型更相似的身份。正 TF 评分表明给定的 TF 应下调以获得与靶细胞类型更相似的身份。
4.细胞命运分类和免疫浸润比较
- 免疫浸润评估主要是指在特定组织(如肿瘤组织)中,不同类型的免疫细胞的存在与活性的分析。这涉及到分析如何及在什么程度上各种免疫细胞(例如
T细胞、B细胞、巨噬细胞等)参与到组织的免疫应答中。免疫浸润的水平可以作为疾病预后的一个重要指标,特别是在癌症研究中,高水平的免疫浸润通常与较好的预后相关。 - 拿常见的CIBERSORT来看,这是用于从复杂的组织表达数据中,通过特征矩阵例如LM22估计细胞组成的相对丰度的免疫浸润方法。
- PACNet是用于分析特别是在癌症研究中常见的多组分样本(如肿瘤微环境中的细胞)。它提供了一种网络方法,通过整合表达数据和先验分子网络信息,来预测样本中细胞类型的丰度。
- 各有侧重,CIBERSORT 更专注于从复杂组织样本中准确估计免疫细胞的丰度,而 PACNet 则提供了一种网络分析方法,不仅可以估计细胞丰度,还可以探究细胞之间的相互作用和网络结构。
对于做分化、做干细胞、做肿瘤分型等来说,这真是一大利器,埋下伏笔,下期更新单细胞的~
相关文章:
PACNet CellNet(代码开源)|bulk数据作细胞分类,评估细胞命运性能的一大利器
文章目录 1.前言2.CellNet2.1CellNet简介2.2CellNet结果 3.PACNet3.1安装R包与加载R包3.2加载数据3.3开始训练和分类3.4可视化分类过程3.5可视化分类结果 4.细胞命运分类和免疫浸润比较 1.前言 今天冲浪看到一个细胞分类性能评估的R包——PACNet,它与转录组分析方法…...
 Object Pascal 学习笔记---第10章第1节(定义属性))
(delphi11最新学习资料) Object Pascal 学习笔记---第10章第1节(定义属性)
第10章 属性和事件 在过去的三章中,我已经介绍了Object Pascal中面向对象编程(OOP)的基础知识,解释了这些概念并展示了大多数面向对象编程语言中通用特性是如何具体实现的。自Delphi的早期,Object Pascal语言就是一…...
【网络安全 | 密码学】JWT基础知识及攻击方式详析
前言 JWT(Json Web Token)是一种用于在网络应用之间安全地传输信息的开放标准。它通过将用户信息以JSON格式加密并封装在一个token中,然后将该token发送给服务端进行验证,从而实现身份验证和授权。 流程 JWT的加密和解密过程如…...
Chrome修改主题颜色
注意:自定义Chrome按钮只在搜索引擎为Google的时候出现。...

大数据:【学习笔记系列】Flink基础架构
Apache Flink 是一个开源的流处理框架,用于处理有界和无界的数据流。Flink 设计用于运行在所有常见的集群环境中,并且能够以高性能和可扩展的方式进行实时数据处理和分析。下面将详细介绍 Flink 的基础架构组件和其工作原理。 1. Flink 架构概览 Flink…...

Debezium系列之:部署Debezium采集Oracle数据库的详细步骤
Debezium系列之:部署Debezium采集Oracle数据库的详细步骤 一、部署Debezium Oracle连接器二、Debezium Oracle 连接器配置三、添加连接器配置四、可插拔数据库与不可插拔数据库一、部署Debezium Oracle连接器 部署的详细步骤可以参考博主这篇技术文章: Debezium系列之:安装…...

C语言通过键盘输入给结构体内嵌的结构体赋值——指针法
1 需求 以录入学生信息(姓名、学号、性别、出生日期)为例,首先通过键盘输入需要录入的学生的数量,再依次输入这些学生的信息,输入完成后输出所有信息。 2 代码 #include<stdio.h> #include<stdlib.h>//…...

AWS Key disabler:AWS IAM用户访问密钥安全保护工具
关于AWS Key disabler AWS Key disabler是一款功能强大的AWS IAM用户访问密钥安全保护工具,该工具可以通过设置一个时间定量来禁用AWS IAM用户访问密钥,以此来降低旧访问密钥所带来的安全风险。 工具运行流程 AWS Key disabler本质上是一个Lambda函数&…...
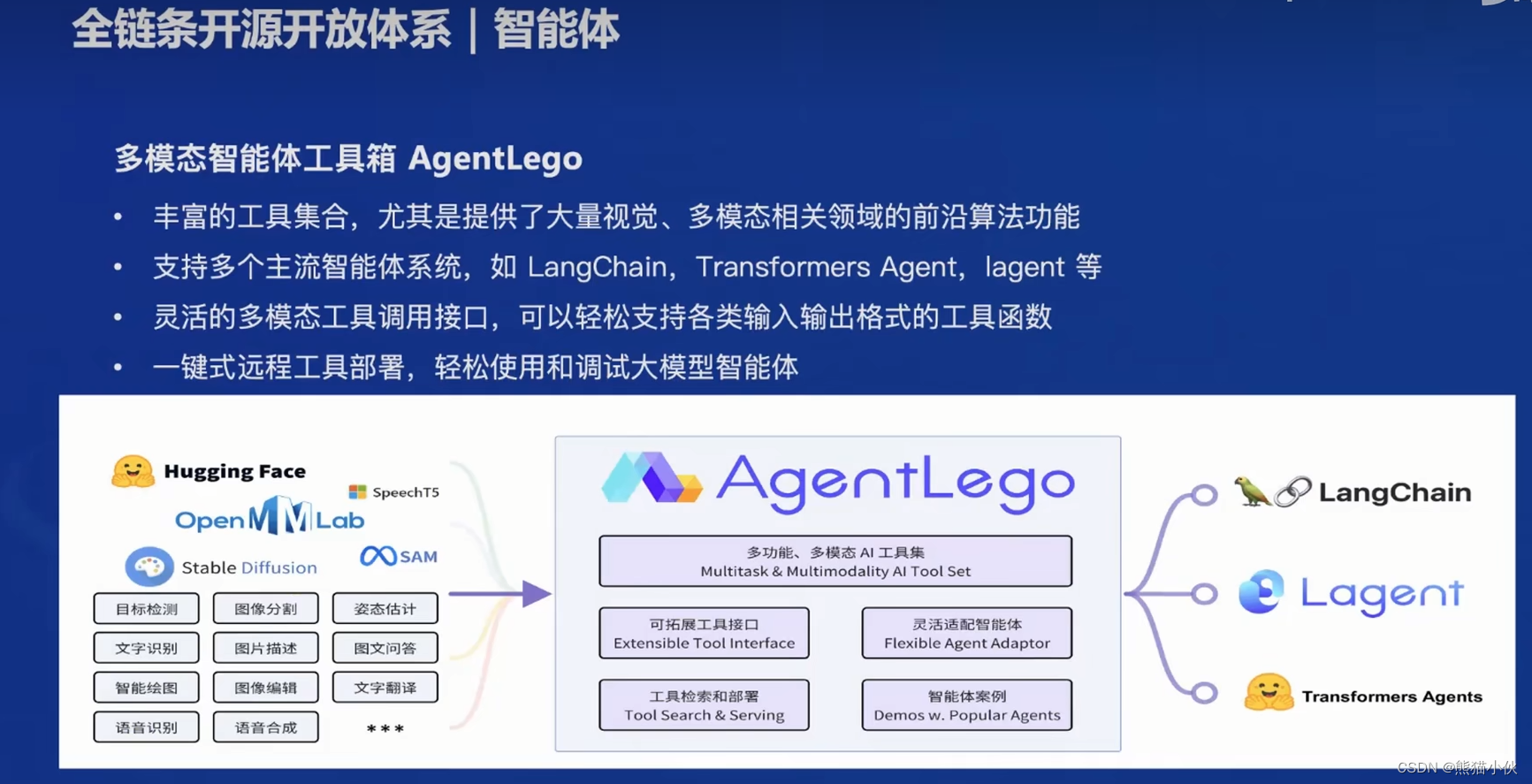
【第1节】书生·浦语大模型全链路开源开放体系
目录 1 简介2 内容(1)书生浦语大模型发展历程(2)体系(3)亮点(4)全链路体系构建a.数据b 预训练c 微调d 评测e.模型部署f.agent 智能体 3 相关论文解读4 ref 1 简介 书生浦语 InternLM…...

代码随想录-链表 | 707设计链表
代码随想录-数组 | 707设计链表 LeetCode 707-设计链表解题思路代码复杂度难点总结 LeetCode 707-设计链表 题目链接 题目描述 你可以选择使用单链表或者双链表,设计并实现自己的链表。 单链表中的节点应该具备两个属性:val 和 next 。val 是当前节点…...
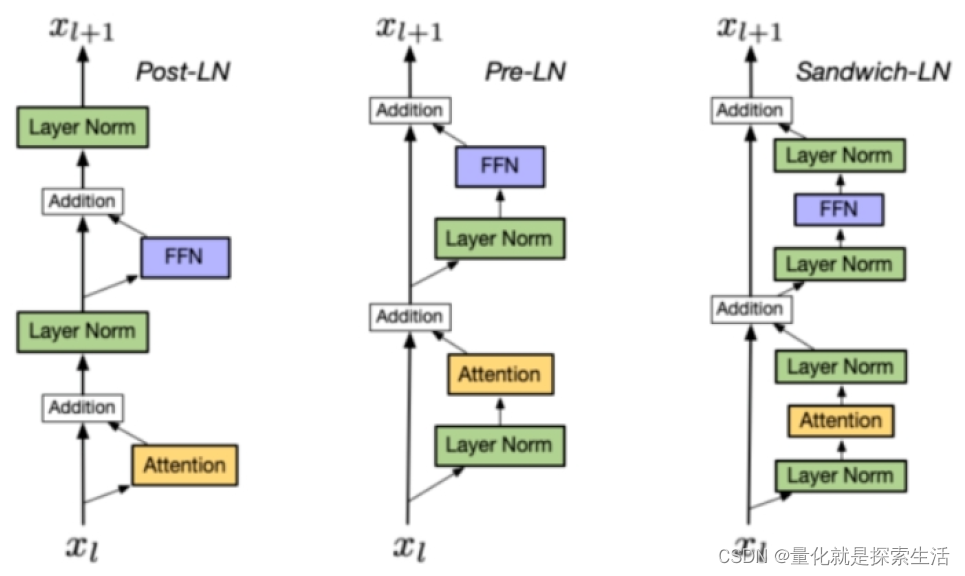
AIGC算法1:Layer normalization
1. Layer Normalization μ E ( X ) ← 1 H ∑ i 1 n x i σ ← Var ( x ) 1 H ∑ i 1 H ( x i − μ ) 2 ϵ y x − E ( x ) Var ( X ) ϵ ⋅ γ β \begin{gathered}\muE(X) \leftarrow \frac{1}{H} \sum_{i1}^n x_i \\ \sigma \leftarrow \operatorname{Var}(…...

【C语言】——字符串函数的使用与模拟实现(下)
【C语言】——字符串函数的使用与模拟实现(下) 前言五、长度受限类字符串函数5.1、 s t r n c p y strncpy strncpy 函数5.2、 s t r n c a t strncat strncat 函数5.3、 s t r n c m p strncmp strncmp 函数 六、 s t r s t r strstr strstr 函数6.1、函…...

mac安装nvm详细教程
0. 前提 清除电脑上原有的node (没有装过的可以忽略)1、首先查看电脑上是否安装的有node,查看node版本node -v2、如果有node就彻底删除nodesudo rm -rf /usr/local/{bin/{node,npm},lib/node_modules/npm,lib/node,share/man/*/node.*}2、保证自己的电脑上有安装git,不然下载n…...

上线流程及操作
上节回顾 1 搜索功能-前端:搜索框,搜索结果页面-后端:一种类型课程-APIResponse(actual_courseres.data.get(results),free_course[],light_course[])-搜索,如果数据量很大,直接使用mysql,效率非常低--》E…...

MobX入门指南:快速上手状态管理库
一、什么是MobX MobX 是一个状态管理库,它可以让你轻松地管理应用程序的状态,并且可以扩展和维护。它使用观察者模式来自动传播你的状态的变化到你的 React 组件。 二、安装及配置 安装 MobX 和 MobX-React:你可以使用 npm 或 yarn 安装这…...

技术洞察:Selenium WebDriver中Chrome, Edge, 和IE配置的关键区别
综述 webdriver.EdgeOptions(), webdriver.ChromeOptions(), 和 webdriver.IeOptions() 都是 Selenium WebDriver 的配置类,用于定制化启动各自浏览器的设置。它们分别对应 Microsoft Edge,Google Chrome,和 Internet Explorer 浏览器。 每…...

使用自定义OCR提升UIE-X检测效果:结合PaddleOCR和UIE模型进行文档信息提取
在实际应用中,识别文档中的特定信息对于许多任务至关重要,例如发票识别、表格信息提取等。然而,由于文档的多样性和复杂性,传统的光学字符识别(OCR)技术可能无法准确识别文档中的信息。为了解决这个问题&am…...

题目:写一个函数,求一个字符串的长度,在main函数中输入字符串,并输出其长度。
题目:写一个函数,求一个字符串的长度,在main函数中输入字符串,并输出其长度。 There is no nutrition in the blog content. After reading it, you will not only suffer from malnutrition, but also impotence. The blog con…...
.net反射(Reflection)
文章目录 一.概念:二.反射的作用:三.代码案例:四.运行结果: 一.概念: .NET 反射(Reflection)是指在运行时动态地检查、访问和修改程序集中的类型、成员和对象的能力。通过反射,你可…...

P1278 单词游戏 简单搜索+玄学优化
单词游戏 传送门 题目描述 Io 和 Ao 在玩一个单词游戏。 他们轮流说出一个仅包含元音字母的单词,并且后一个单词的第一个字母必须与前一个单词的最后一个字母一致。 游戏可以从任何一个单词开始。 任何单词禁止说两遍,游戏中只能使用给定词典中含有…...
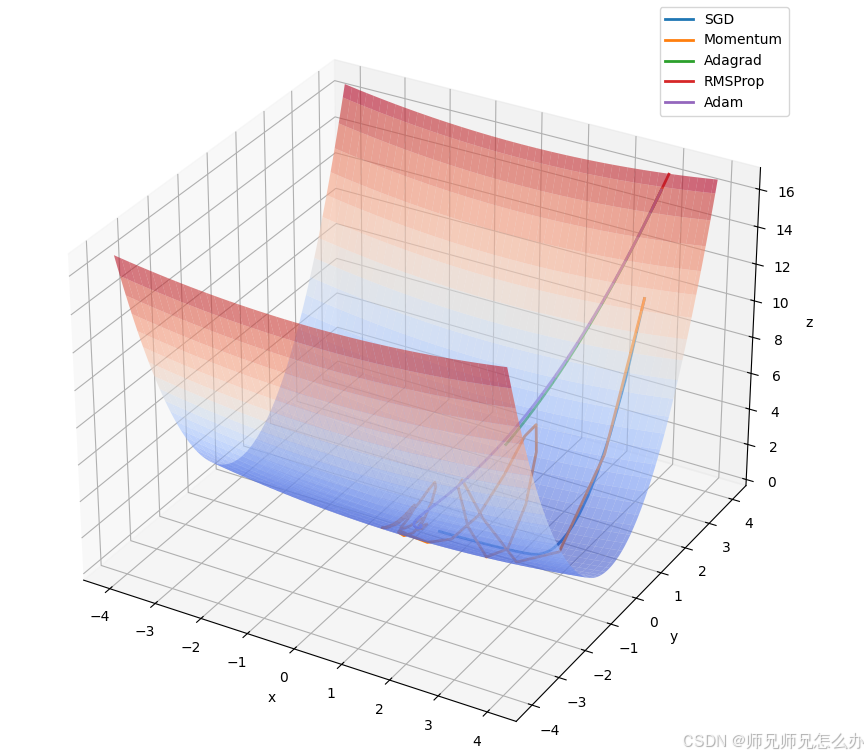
【人工智能】神经网络的优化器optimizer(二):Adagrad自适应学习率优化器
一.自适应梯度算法Adagrad概述 Adagrad(Adaptive Gradient Algorithm)是一种自适应学习率的优化算法,由Duchi等人在2011年提出。其核心思想是针对不同参数自动调整学习率,适合处理稀疏数据和不同参数梯度差异较大的场景。Adagrad通…...
)
Spring Boot 实现流式响应(兼容 2.7.x)
在实际开发中,我们可能会遇到一些流式数据处理的场景,比如接收来自上游接口的 Server-Sent Events(SSE) 或 流式 JSON 内容,并将其原样中转给前端页面或客户端。这种情况下,传统的 RestTemplate 缓存机制会…...

Cesium1.95中高性能加载1500个点
一、基本方式: 图标使用.png比.svg性能要好 <template><div id"cesiumContainer"></div><div class"toolbar"><button id"resetButton">重新生成点</button><span id"countDisplay&qu…...
python/java环境配置
环境变量放一起 python: 1.首先下载Python Python下载地址:Download Python | Python.org downloads ---windows -- 64 2.安装Python 下面两个,然后自定义,全选 可以把前4个选上 3.环境配置 1)搜高级系统设置 2…...
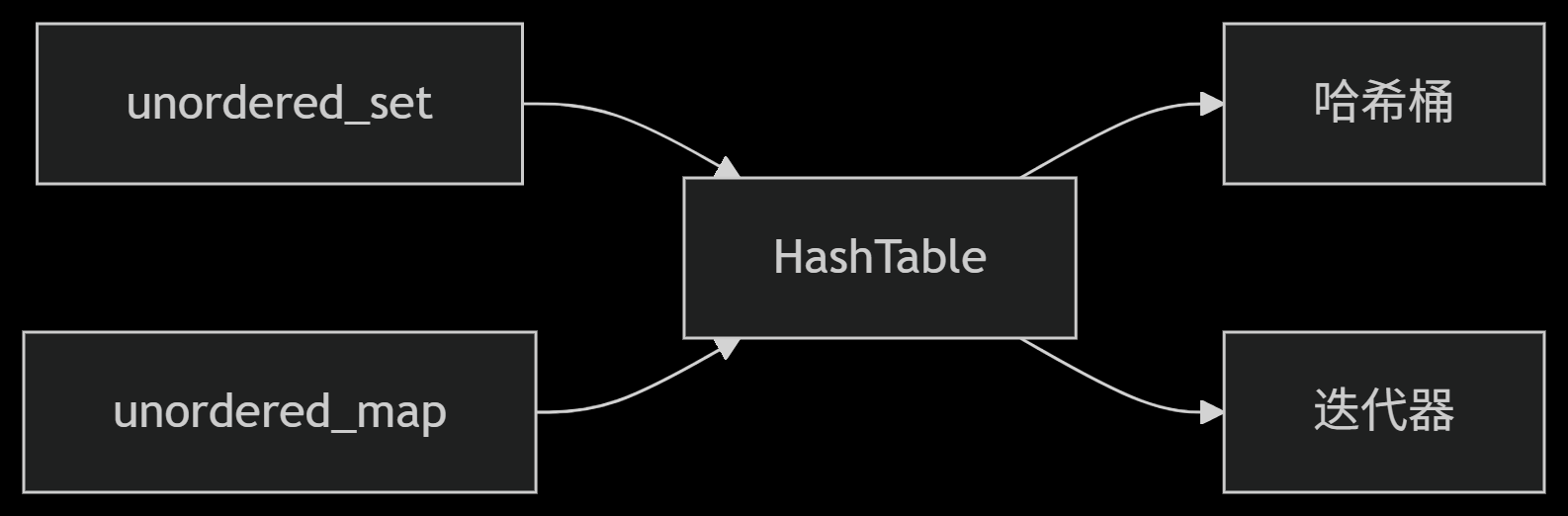
从零实现STL哈希容器:unordered_map/unordered_set封装详解
本篇文章是对C学习的STL哈希容器自主实现部分的学习分享 希望也能为你带来些帮助~ 那咱们废话不多说,直接开始吧! 一、源码结构分析 1. SGISTL30实现剖析 // hash_set核心结构 template <class Value, class HashFcn, ...> class hash_set {ty…...
基础光照(Basic Lighting))
C++.OpenGL (10/64)基础光照(Basic Lighting)
基础光照(Basic Lighting) 冯氏光照模型(Phong Lighting Model) #mermaid-svg-GLdskXwWINxNGHso {font-family:"trebuchet ms",verdana,arial,sans-serif;font-size:16px;fill:#333;}#mermaid-svg-GLdskXwWINxNGHso .error-icon{fill:#552222;}#mermaid-svg-GLd…...

UR 协作机器人「三剑客」:精密轻量担当(UR7e)、全能协作主力(UR12e)、重型任务专家(UR15)
UR协作机器人正以其卓越性能在现代制造业自动化中扮演重要角色。UR7e、UR12e和UR15通过创新技术和精准设计满足了不同行业的多样化需求。其中,UR15以其速度、精度及人工智能准备能力成为自动化领域的重要突破。UR7e和UR12e则在负载规格和市场定位上不断优化…...
C# 求圆面积的程序(Program to find area of a circle)
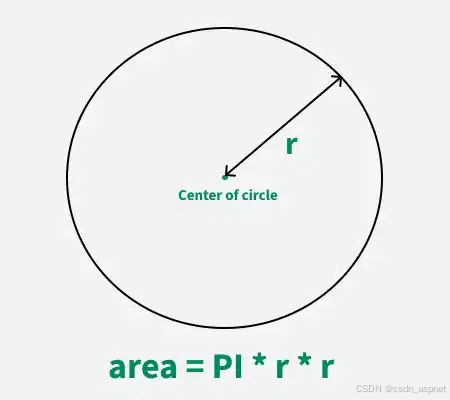
给定半径r,求圆的面积。圆的面积应精确到小数点后5位。 例子: 输入:r 5 输出:78.53982 解释:由于面积 PI * r * r 3.14159265358979323846 * 5 * 5 78.53982,因为我们只保留小数点后 5 位数字。 输…...

用机器学习破解新能源领域的“弃风”难题
音乐发烧友深有体会,玩音乐的本质就是玩电网。火电声音偏暖,水电偏冷,风电偏空旷。至于太阳能发的电,则略显朦胧和单薄。 不知你是否有感觉,近两年家里的音响声音越来越冷,听起来越来越单薄? —…...

安宝特方案丨船舶智造的“AR+AI+作业标准化管理解决方案”(装配)
船舶制造装配管理现状:装配工作依赖人工经验,装配工人凭借长期实践积累的操作技巧完成零部件组装。企业通常制定了装配作业指导书,但在实际执行中,工人对指导书的理解和遵循程度参差不齐。 船舶装配过程中的挑战与需求 挑战 (1…...