基于qiime2的16S数据分析全流程:从导入数据到下游分析一条龙
目录
创建metadata
把数据导入qiime2
去除引物序列
双端合并 (dada2不需要)
质控 (dada2不需要)
使用deblur获得特征序列
使用dada2生成代表序列与特征表
物种鉴定
可视化物种鉴定结果
构建进化树(ITS一般不构建进化树)
生成多样性分析所需数据
α多样性分析
beta多样性分析
绘制稀释曲线
差异物种分析
如何把.qza格式文件导出?比如把特征表导出
演示单端数据导入qiime2


创建metadata


把数据导入qiime2
去除引物序列
双端合并 (dada2不需要)
质控 (dada2不需要)
使用deblur获得特征序列

使用dada2生成代表序列与特征表
物种鉴定
可视化物种鉴定结果


构建进化树(ITS一般不构建进化树)


生成多样性分析所需数据


α多样性分析

beta多样性分析

绘制稀释曲线

差异物种分析
如何把.qza格式文件导出?比如把特征表导出
演示单端数据导入qiime2

相关文章:
基于qiime2的16S数据分析全流程:从导入数据到下游分析一条龙
目录 创建metadata 把数据导入qiime2 去除引物序列 双端合并 (dada2不需要) 质控 (dada2不需要) 使用deblur获得特征序列 使用dada2生成代表序列与特征表 物种鉴定 可视化物种鉴定结果 构建进化树(ITS一般不构建进化树…...
)
【软件测试开发】:软件测试常用函数1.0(C++)
1. 元素的定位 web⾃动化测试的操作核⼼是能够找到⻚⾯对应的元素,然后才能对元素进⾏具体的操作。 常⻅的元素定位⽅式⾮常多,如id,classname,tagname,xpath,cssSelector 常⽤的主要由cssSelector和xpath…...

vue2项目修改浏览器显示的网页图标
1.准备一个新的图标文件,通常是. ico格式,也可以是. Png、. Svg等格式 2.将新的图标文件(例如:faviconAt.png)放入项目的public文件夹中。如下图 public文件夹中的所有文件都会在构建时原样复制到最终的输出目录(通常是dist) 3. 修改vue项目…...

开源、创新与人才发展:机器人产业的战略布局与稚晖君成功案例解析
目录 引言 一、开源:机器人产业的战略布局 促进技术进步和生态建设 吸引人才和合作伙伴 建立标准和网络效应 降低研发风险与成本 二、稚晖君:华为"天才少年计划"的成功典范 深厚的技术积累与动手能力 强烈的探索和创新意识 持续公开…...

线程相关作业
1.创建两个线程,分支线程1拷贝文件的前一部分,分支线程2拷贝文件的后一部分 #include "head.h"#define BUFFER_SIZE 1024// 线程参数结构体,包含文件名和文件偏移量 typedef struct {FILE *src_file;FILE *dest_file;long start_o…...

通义万相2.1开源版本地化部署攻略,生成视频再填利器
2025 年 2 月 25 日晚上 11:00 通义万相 2.1 开源发布,前两周太忙没空搞它,这个周末,也来本地化部署一个,体验生成效果如何,总的来说,它在国内文生视频、图生视频的行列处于领先位置,…...

【模拟CMOS集成电路设计】带隙基准(Bandgap)设计与仿真(基于运放的电流模BGR)
【模拟CMOS集成电路设计】带隙基准(Bandgap)设计与仿真 前言工程文件&部分参数计算过程,私聊~ 一、 设计指标指标分析: 二、 电路分析三、 仿真3.1仿真电路图3.2仿真结果(1)运放增益(2)基准温度系数仿真(3)瞬态启动仿真(4)静态…...

如何选择国产串口屏?
目录 1、迪文 2、淘晶驰 3、广州大彩 4、金玺智控 5、欣瑞达 6、富莱新 7、冠显 8、有彩 串口屏,顾名思义,就是通过串口通信接口(如RS232、RS485、TTL UART等)与主控设备进行通信的显示屏。其核心功能是显示信息和接收输入…...

Solana中的程序派生地址(PDAs):是什么,为什么,以及如何?
程序派生地址 (PDA) 在 Solana 中的应用:什么、为什么和如何? 在学习 Solana 时,你会经常听到关于 程序派生地址 (PDAs) 的讨论。它们就像这样 —— 强大、多功能,而且最重要的是,稍微被误解。如果你是一个开发者&…...

利用FatJar彻底解决Jar包冲突(一)
利用FatJar彻底解决Jar包冲突 序FatJar的加载与隔离⼀、 FatJar概念⼆、FatJar的加载三、FatJar的隔离四、隔离机制验证五、 FatJar的定位六、 打包注意点 序 今天整理旧电脑里的资料,偶然翻到大概10年前实习时写的笔记,之前经常遇到Java依赖冲突的问题…...

Spring MVC笔记
01 什么是Spring MVC Spring MVC 是 Spring 框架中的一个核心模块,专门用于构建 Web 应用程序。它基于经典的 MVC 设计模式(Model-View-Controller),但通过 Spring 的特性(如依赖注入、注解驱动)大幅简化了…...
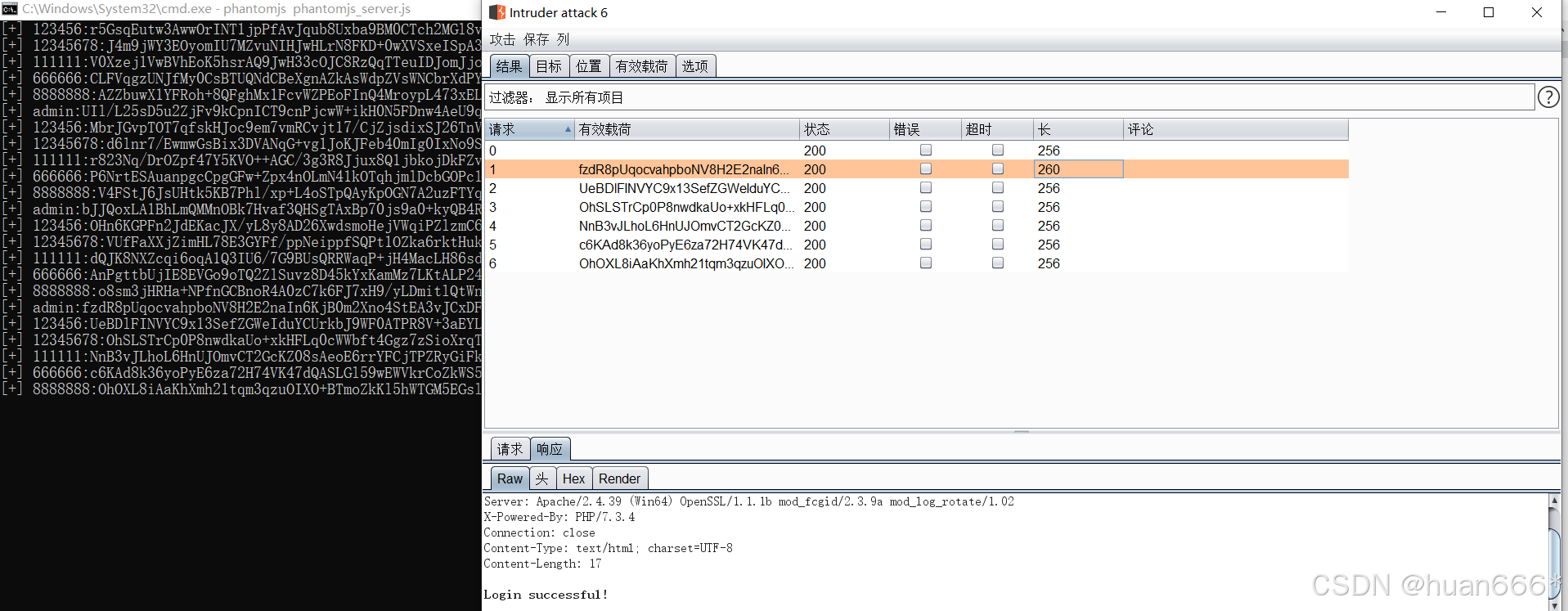
BurpSuite插件jsEncrypter使用教程
一、前言 在当今Web应用安全测试中,前端加密已成为开发者保护敏感数据的常用手段。然而,这也给安全测试人员带来了挑战,传统的抓包方式难以获取明文数据,测试效率大打折扣。BurpSuite作为一款强大的Web安全测试工具,其…...

【C#实现手写Ollama服务交互,实现本地模型对话】
前言 C#手写Ollama服务交互,实现本地模型对话 最近使用C#调用OllamaSharpe库实现Ollama本地对话,然后思考着能否自己实现这个功能。经过一番查找,和查看OllamaSharpe源码发现确实可以。其实就是开启Ollama服务后,发送HTTP请求&a…...

Android Glide 框架线程管理模块原理的源码级别深入分析
一、引言 在现代的 Android 应用开发中,图片加载是一个常见且重要的功能。Glide 作为一款广泛使用的图片加载框架,以其高效、灵活和易用的特点受到了开发者的青睐。其中,线程管理模块是 Glide 框架中至关重要的一部分,它负责协调…...

每天记录一道Java面试题---day32
MySQL索引的数据结构、各自优劣 回答重点 B树:是一个平衡的多叉树,从根节点到每个叶子节点的高度差不超过1,而且同层级的节点间有指针相互连接。在B树上的常规检索,从根节点到叶子节点的搜索效率基本相当,不会出现大…...

Vue3 Pinia 符合直觉的Vue.js状态管理库
Pinia 符合直觉的Vue.js状态管理库 什么时候使用Pinia 当两个关系非常远的组件,要传递参数时使用Pinia组件的公共参数使用Pinia...

深度学习与大模型基础-向量
大家好!今天我们来聊聊向量(Vector)。别被这个词吓到,其实向量在我们的生活中无处不在,只是我们没注意罢了。 1. 向量是什么? 简单来说,向量就是有大小和方向的量。比如你从家走到学校&#x…...

【网络编程】完成端口 IOCP
10.11 完成端口 10.11.1 基本概念 完成端口的全称是I/O 完成端口,英文为IOCP(I/O Completion Port) 。IOCP是一个异 步I/O 的 API, 可以高效地将I/O 事件通知给应用程序。与使用select() 或是其他异步方法不同 的是,一个套接字与一个完成端口关联了起来…...

《苍穹外卖》SpringBoot后端开发项目重点知识整理(DAY1 to DAY3)
目录 一、在本地部署并启动Nginx服务1. 解压Nginx压缩包2. 启动Nginx服务3. 验证Nginx是否启动成功: 二、导入接口文档1. 黑马程序员提供的YApi平台2. YApi Pro平台3. 推荐工具:Apifox 三、Swagger1. 常用注解1.1 Api与ApiModel1.2 ApiModelProperty与Ap…...

管理网络安全
防火墙在 Linux 系统安全中有哪些重要的作用? 防火墙作为网络安全的第一道防线,能够根据预设的规则,对进出系统的网络流量进行严格筛选。它可以阻止未经授权的外部访问,只允许符合规则的流量进入系统,从而保护系统免受…...
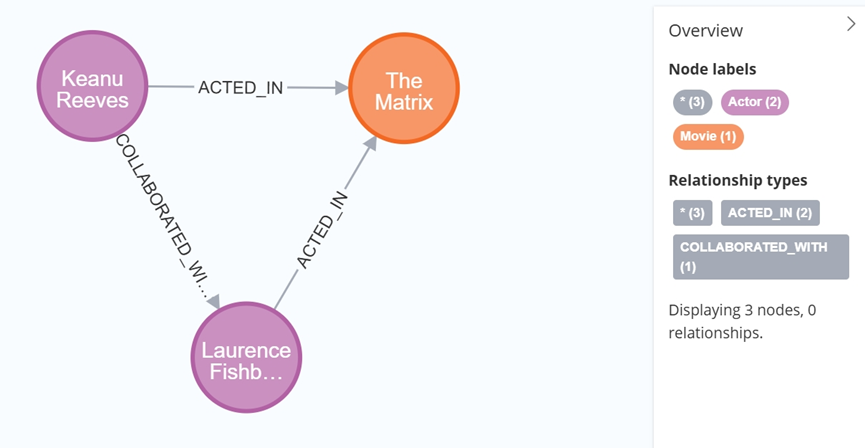
大数据学习栈记——Neo4j的安装与使用
本文介绍图数据库Neofj的安装与使用,操作系统:Ubuntu24.04,Neofj版本:2025.04.0。 Apt安装 Neofj可以进行官网安装:Neo4j Deployment Center - Graph Database & Analytics 我这里安装是添加软件源的方法 最新版…...

QMC5883L的驱动
简介 本篇文章的代码已经上传到了github上面,开源代码 作为一个电子罗盘模块,我们可以通过I2C从中获取偏航角yaw,相对于六轴陀螺仪的yaw,qmc5883l几乎不会零飘并且成本较低。 参考资料 QMC5883L磁场传感器驱动 QMC5883L磁力计…...
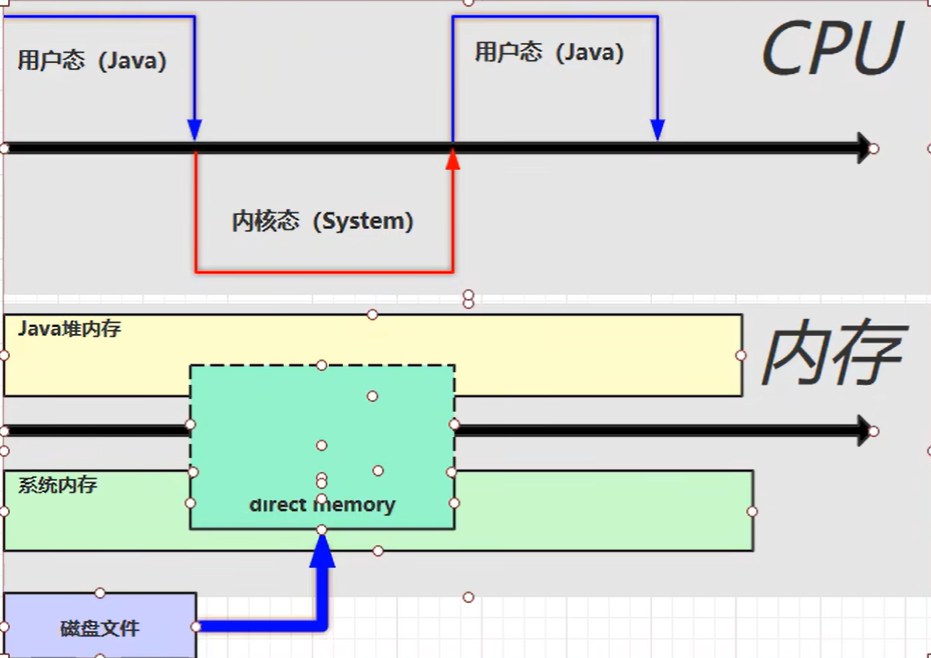
【JVM】- 内存结构
引言 JVM:Java Virtual Machine 定义:Java虚拟机,Java二进制字节码的运行环境好处: 一次编写,到处运行自动内存管理,垃圾回收的功能数组下标越界检查(会抛异常,不会覆盖到其他代码…...

2024年赣州旅游投资集团社会招聘笔试真
2024年赣州旅游投资集团社会招聘笔试真 题 ( 满 分 1 0 0 分 时 间 1 2 0 分 钟 ) 一、单选题(每题只有一个正确答案,答错、不答或多答均不得分) 1.纪要的特点不包括()。 A.概括重点 B.指导传达 C. 客观纪实 D.有言必录 【答案】: D 2.1864年,()预言了电磁波的存在,并指出…...
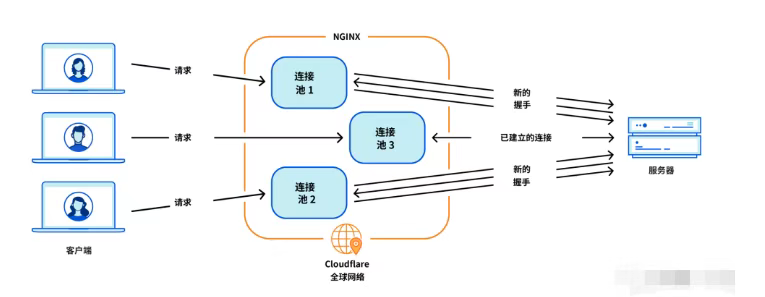
Cloudflare 从 Nginx 到 Pingora:性能、效率与安全的全面升级
在互联网的快速发展中,高性能、高效率和高安全性的网络服务成为了各大互联网基础设施提供商的核心追求。Cloudflare 作为全球领先的互联网安全和基础设施公司,近期做出了一个重大技术决策:弃用长期使用的 Nginx,转而采用其内部开发…...

Caliper 配置文件解析:config.yaml
Caliper 是一个区块链性能基准测试工具,用于评估不同区块链平台的性能。下面我将详细解释你提供的 fisco-bcos.json 文件结构,并说明它与 config.yaml 文件的关系。 fisco-bcos.json 文件解析 这个文件是针对 FISCO-BCOS 区块链网络的 Caliper 配置文件,主要包含以下几个部…...
)
Android第十三次面试总结(四大 组件基础)
Activity生命周期和四大启动模式详解 一、Activity 生命周期 Activity 的生命周期由一系列回调方法组成,用于管理其创建、可见性、焦点和销毁过程。以下是核心方法及其调用时机: onCreate() 调用时机:Activity 首次创建时调用。…...

华硕a豆14 Air香氛版,美学与科技的馨香融合
在快节奏的现代生活中,我们渴望一个能激发创想、愉悦感官的工作与生活伙伴,它不仅是冰冷的科技工具,更能触动我们内心深处的细腻情感。正是在这样的期许下,华硕a豆14 Air香氛版翩然而至,它以一种前所未有的方式&#x…...
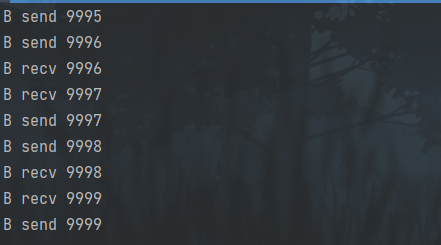
并发编程 - go版
1.并发编程基础概念 进程和线程 A. 进程是程序在操作系统中的一次执行过程,系统进行资源分配和调度的一个独立单位。B. 线程是进程的一个执行实体,是CPU调度和分派的基本单位,它是比进程更小的能独立运行的基本单位。C.一个进程可以创建和撤销多个线程;同一个进程中…...
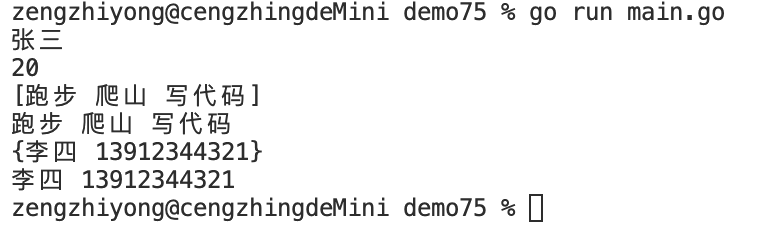
Golang——7、包与接口详解
包与接口详解 1、Golang包详解1.1、Golang中包的定义和介绍1.2、Golang包管理工具go mod1.3、Golang中自定义包1.4、Golang中使用第三包1.5、init函数 2、接口详解2.1、接口的定义2.2、空接口2.3、类型断言2.4、结构体值接收者和指针接收者实现接口的区别2.5、一个结构体实现多…...