单细胞多组学整合与对齐的计算方法
Computational Methods for Single-cell Multi-omics Integration and Alignment
Bioinformatics-2022-密西根大学
关键词:单细胞;多组学;机器学习;无监督学习;集成
摘要
最近发展起来的生成单细胞基因组数据的技术在生物学领域产生了革命性的影响。多组学测定提供了更大的机会来理解细胞状态和生物过程。然而,整合具有非常不同维度和统计特性的不同组学数据的问题仍然非常具有挑战性。为此任务正在开发越来越多的计算工具,利用了从机器翻译到网络理论的思想,代表着生物学和数据科学交叉口上的又一个前沿。我们在这篇综述中的目标是全面、及时地调查用于整合单细胞多组学数据的计算技术,同时使每种算法背后的概念易于理解,适用于非专业人士。
单模态测序手段,对一个单细胞只能测定一种表达:包括scRNA-seq(单细胞转录组), scATAC-seq(染色质可及性), single-cell bisulfite sequencing(DNA甲基化)。通过假设通过不同技术检测的细胞具有相似的特性,可以使用比对方法在不同的组学检测中计算聚集相似的细胞,并得出一致的生物学推断---对齐任务
多模态测序手段,对一个单细胞同时测定多种表达: CITE-seq(转录组和蛋白质)、 scG&T-seq(单细胞基因组和转录组测序)、scM&T-seq(单细胞甲基组和转录组测序) 、SHARE-seq(单细胞染色质和转录组数据)、SNARE-seq(单核染色质可及性和mRNA表达测序)、scTrioseq(单核苷酸多态性(SNPs)、基因表达和DNA甲基)、scNMT(转录组、染色质可及性和DNA甲基化) ---整合分析任务、对齐任务
引言
单细胞测序技术以前所未有的高分辨率探索了生物过程的契机。诸如Drop-seq [1]、InDrops [2]和10x Genomics assays [3]等技术能够同时测量成千上万个单细胞的基因表达[单细胞RNA测序(scRNA-seq)]。其他数据模式的测量也日益可用。例如,单细胞转座酶可及染色质测序(scATAC-seq)评估染色质可及性,而单细胞亚硫酸盐测序捕获单细胞的DNA甲基化。然而,许多这类技术设计用于测量单一模态,不适合进行多组学测量。因此,合并来自这些测量的信息的方式是从同一样本的不同子集中检测不同组学。通过假设由不同技术检测的细胞具有相似的特性,可以使用对齐方法在计算上整合不同组学测量中的相似细胞,并得出共识的生物推论。
最近,然而,已经开发出了一些实验技术,能够同时从同一组单个细胞中测定多种模态。细胞转录组和表位的测序(CITE-seq)[4] 和RNA表达和蛋白质测序(REAP-seq)[5] 可以测量蛋白质和基因表达。单核染色质可及性和mRNA表达测序(SNARE-seq)[5,6],具有测序的同时高通量ATAC和RNA表达(SHARE-seq)[7],以及染色质可及性和mRNA的单细胞组合分析(sci-CAR)[8] 可以测量基因表达和染色质可及性,而基因表达和甲基化的单细胞测序(scGEM)[9] 可以测量基因表达和DNA甲基化,基因组和转录组测序(G&T-seq)[10] 可以测定基因组和转录组。对于三重组学数据生成,单细胞核小体、甲基化和转录测序(scNMT)[11] 可以测量基因表达、染色质可及性和DNA甲基化,而单细胞三重组学测序(scTrio-seq)[9,12] 可以同时捕获单核苷酸多态性(SNPs)、基因表达和DNA甲基化。10x Genomics 的多组测定平台能够同时测量基因表达和染色质可及性。由于测定中的噪音和稀疏性,以及不同模态的不同统计分布等多种原因,从相同细胞获取的这些数据的综合分析仍然是一项具有挑战性的计算任务。为了明晰起见,我们区分了将来自同一组单个细胞的多组学数据进行综合的方法,与专为处理来自同一组织但不同细胞的多模态数据而设计的对齐方法。它们的差异在图1中显示。
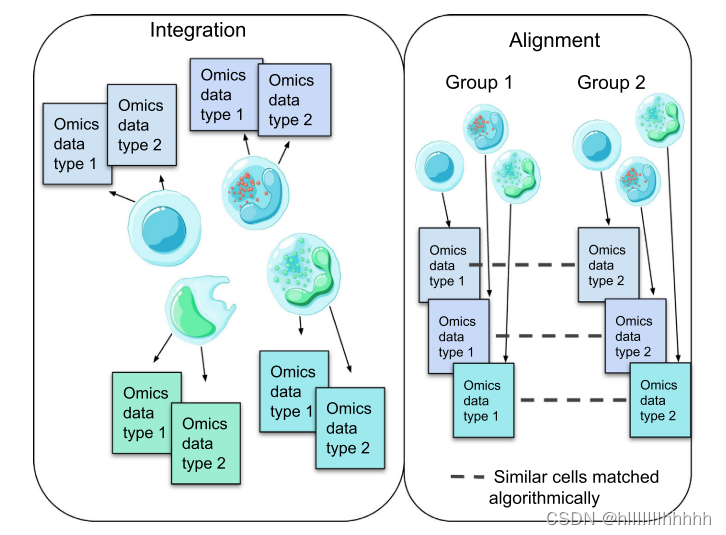
图1 多组学数据的整合与对齐
多组学数据有时可以从相同的单个细胞集中测定(左侧);而有时仅可用来自相同/相似样本但不同单个细胞的测定数据(右侧)。在前一种情况下,我们面临着整合不同数据模态的任务(左侧);而在后一种情况下,我们首先需要在样本之间识别相似的细胞(右侧)。这是对齐的计算任务。
右侧图:从相同组织的相似但不同的细胞中获得的组学数据,基于对齐算法,得到单个细胞下的两种数据信息
多组学测序数据的数据融合算法应用早于单细胞技术;在先前的综述中,已经使用了各种计算工具对批量水平的数据进行了整合[13]。在本综述中,我们旨在为计算生物学领域的研究人员提供单细胞领域中现有的多组学数据整合和对齐的计算工具的全面、最新总结。对于更一般的调查,鼓励读者查阅其他单细胞多组学综述[14–21]。与众不同的是,我们综述的目标读者是试图在详细的技术层面了解计算工具的计算生物学家。因此,这里的工作深入介绍了底层算法的基本原理,并在适用时详细阐述了这些方法的优势和劣势。
处理从相同单细胞生成的多组学数据的集成方法
从同一组单细胞分析的多模态数据的整合方法可以被概念化为“垂直整合”,这在早期的综述[17]中提到,可以通过方法学大致分为至少三种主要类型:基于数学矩阵因式分解的方法,人工智能(AI;例如,基于神经网络)的方法和基于网络的方法。这些方法的方案如图2所示。其他不太多样化的方法包括贝叶斯统计方法和度量学习方法。表1和表2总结了当前实施的方法列表。
矩阵分解方法
基于矩阵分解的方法旨在将每个细胞描述为每个组学元素(基因、表观遗传位点和蛋白质)的向量与捕获其基本特性的缩减和共同特征(因子)的向量的乘积(图2A)。
目标: 这种方法的目标是描述每个细胞,将其表示为每个组学元素(如基因、表观遗传位点和蛋白质)的向量。每个组学元素都可以在一个矩阵中表示,形成一个多组学数据矩阵,其中每行代表一个细胞,每列代表一个组学元素。
原理: 通过矩阵分解,这种方法将整个多组学数据矩阵分解为两个矩阵的乘积。其中一个矩阵表示每个细胞与捕获其基本特性的缩减和共同特征(因子)的向量,而另一个矩阵表示每个组学元素的向量。这样,每个细胞的整体表示可以被分解成对应于每个组学元素的向量的组合。
从数学上讲,如果我们将每个组学表示为矩阵Xi(i=1,2,...),那么矩阵分解将其分解为在所有组学数据类型上共享的矩阵H以及组学特定矩阵Wi(i=1,2,...),再加上随机噪声ei(i=1,2,...),表示为:
![]()

这类方法简单且易于解释,因为细胞和组学因子可以与组学特征关联,但可能缺乏捕捉非线性效应的能力。我们将在下面描述这种类型方法的变化。MOFA+ [22] 是多组学因子分析(MOFA)[22,23] 的续篇。这两项研究执行因子分析,配备了稀疏诱导的贝叶斯元素,包括自动相关性确定 [24]。MOFA+ 在两个视图(对应不同模态)和组别(对应不同实验条件)上集成数据。该模型可以轻松扩展到大型数据集。MOFA+ 被应用于整合使用 scNMT 从小鼠胚胎测定的基因表达、染色质可及性和DNA甲基化数据,以及整合不同实验条件下的多个数据集,而非不同组学。在对小鼠数据集进行因子分析后,最相关的因子与塑造胚胎发育的生物过程相关。MOFA+ 提供了一个优雅而成功的整合通用框架,尽管在特定情况下,它可能会被为整合特定组学层设计的更专业化的模型所取代。
单细胞聚合和推断(scAI)[25] 在矩阵因子分解上有所创新,专门设计用于整合表观遗传学(染色质可及性和DNA甲基化)和转录组数据。它通过在相似的细胞之间聚合(平均)表观遗传数据来解决表观遗传数据的稀疏性。这需要一个细胞之间相似性的概念,该概念作为模型的一部分学习,而不是在整合之前假定的。他们的模型解决了以下优化问题:
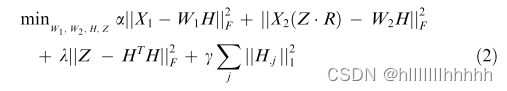
这里,X1代表转录组数据,X2代表表观基因组数据,H是共同的(细胞特定的)因子矩阵,W1和W2是测定特定的因子矩阵,Z是细胞之间相似性矩阵,R的是伯努利分布的随机变量,而超参数a、k和c决定了不同项的相对重要性。对常规矩阵因子分解的创新在于对聚合的表观遗传数据X2(Z・R)进行因子分解,而不是直接对表观遗传数据X2进行因子分解。

学习完成后,细胞因子矩阵用于对细胞进行聚类,并使用载荷矩阵中数值的大小来排名基因和表观遗传标记的重要性。
在上面提到的背景中,学习完成后,细胞因子矩阵 H 被用于对细胞进行聚类,而载荷矩阵 W 中数值的大小则可以用来评估基因和表观遗传标记的重要性。这是因为 W 的数值表示了原始数据中的每个基因或表观遗传标记对于细胞因子的贡献程度。较大的数值表示该基因或标记在形成细胞因子时的重要性较高,从而在细胞之间的差异中起到重要作用。
为了共同可视化不同的因子,scAI实现了一种利用Sammon映射的新型VscAI算法[26]。可以使用相关性分析和非负最小二乘回归来探索表观遗传与基因表达之间的关系。该模型在使用MOSim [27]进行模拟和多个真实数据集上进行测试,相对于先前的MOFA版本,在识别自然簇和将表观遗传数据压缩为有意义因子方面表现更好。
神经网络方法
尽管神经网络通常适用于监督学习任务,但一类名为自编码器的神经网络常用于无监督学习,例如在单细胞中的多组学整合问题。深度自编码器通过将输入通过较低维度的隐藏层(瓶颈)压缩,然后试图将原始输入重构为神经网络的输出,从而执行非线性降维(图2B)。它们由两部分组成:执行降维的编码器网络和根据降维数据进行重构的解码器网络。从原理上讲,自编码器通过允许非线性变换来推广主成分分析。存在许多自编码器模型的变体,其中变分自编码器已被证明对于分析单细胞数据非常有用。变分自编码器不直接在降维的(潜在的)空间中对数据进行编码,而是从潜在空间的概率分布(通常是高斯分布)中进行采样,并使用编码器网络生成该分布的参数。因此,它们将深度学习和贝叶斯推断相结合,生成生成模型,不仅对原始数据进行降维,还产生逼真的合成数据点。在下文中,我们将回顾使用自编码器架构的特定变体来整合单细胞多组学数据的方法。
单细胞多模态变分自编码器(scMVAE)[28] 被设计用于整合转录组和染色质可及性数据,采用了变分自编码器的一种版本。在多组学整合中的关键问题是如何将多组学数据编码成单一的潜在空间表示。在 scMVAE 中,使用了三种不同的方法来完成这个任务,包括一个作用于连接的输入数据的神经网络、在合并之前对转录组和染色质可及性数据分别进行编码的神经网络,以及一种用于组合不同表示的专家乘积技术[29]。同时,用于规范跨细胞表达的细胞特定比例被学习(称为库因子)。通过解码器神经网络处理潜在表示,输入数据通过计算基因缺失的概率并预测被建模为负二项分布的测量基因的表达来进行重构。
Seurat v4 [42] 的目标是将数据表示为一个加权最近邻(WNN)图,其中根据两种模态的一致性相似的细胞被连接起来。在构建 WNN 图的过程中,学习了一组细胞特异性权重,指导不同组学数据的相对重要性。这些权重通常携带重要的生物学含义。具体而言,Seurat v4 管道包括以下步骤:首先,对应于不同组学的数据使用主成分分析(PCA)进行降维到相同的维度数。然后,构建了对应于不同组学的 k-最近邻(kNN)图。在 kNN 图中,每个数据点(该图的节点)与 k 个最近邻节点相连。然后,通过考虑最近邻图的跨模态和内模态预测的准确性来学习确定不同组学相对重要性的细胞特异性系数。最后,在使用前一步中学到的系数的基础上,对来自不同组学的数据进行线性组合。然后,与这些线性组合最近的邻居被连接以构建 WNN 图。Seurat v4 应用于基于 CITE-seq 的转录组和蛋白质组数据集,以及其他涉及 mRNA、蛋白质和染色质可及性的数据集。作者使用与细胞对应的数据与其最近的潜在空间邻居的平均值之间的相关性(Pearson 和 Spearman)比较了该方法与 MOFA+ 和 totalVI,并声称它在这些指标上表现优于 MOFA+ 或 totalVI。
这段话描述了 Seurat v4 方法的主要步骤和应用,梳理如下:
1. **目标:** Seurat v4 的目标是将多组学数据表示为加权最近邻(WNN)图,其中相似的细胞根据两种模态的一致性被连接。
2. **WNN 图构建:**
- 使用主成分分析(PCA),将不同组学的数据降维到相同的维度。
- 构建 k-最近邻(kNN)图,其中每个数据点与 k 个最近邻节点相连。3. **权重学习:**
- 学习细胞特异性权重,这些权重指导了不同组学数据的相对重要性。
- 通过考虑最近邻图的跨模态和内模态预测准确性来确定这些权重。4. **线性组合:**
- 使用前一步学到的权重,对不同组学的数据进行线性组合。5. **WNN 图建立:**
- 将与这些线性组合最近的邻居连接起来,构建 WNN 图。6. **应用:**
- Seurat v4 应用于不同数据集,包括基于 CITE-seq 的转录组和蛋白质组数据,以及涉及 mRNA、蛋白质和染色质可及性的其他数据集。7. **性能比较:**
- 作者使用相关性(Pearson 和 Spearman)比较了 Seurat v4 与 MOFA+ 和 totalVI 方法的性能,声称在这些指标上表现优于 MOFA+ 或 totalVI。
其他模型
BREM-SC [43] 是一种贝叶斯混合方法。它通过将单细胞基因表达和蛋白质数据建模为共享相同基础参数的概率分布混合体来整合它们。该模型可用于执行联合聚类,其中可以通过后验概率来量化对聚类分配的信心度。与单组学聚类方法相比,它表现出色。尽管用于训练模型的马尔可夫链蒙特卡洛(MCMC)过程可能计算密集,但该模型通过利用概率分布来考虑两个组学层之间的差异,提供了一种有效的整合方式。
SCHEMA [44] 是一种不同的度量学习方法,旨在构建样本空间上的距离概念,考虑了不同的组学数据。其中的一种组学(通常是单细胞RNA测序)被视为距离的主要基础,然后使用其他组学来修改这个距离。这被制定为使用二次规划优化二次函数。因此,可以整合单细胞RNA测序和单细胞ATAC测序数据,从而为细胞发育轨迹提供深层次的见解。与基于分别对不同模态进行聚类或使用规范相关分析进行整合的方法相比,该方法展现出更好的聚类性能。它是一种用于非对称整合不同质量数据模态的有用方法,例如单细胞RNA测序和单细胞ATAC测序数据的情况。
处理来自同一组织的不同单个细胞产生的多组基因组数据的对齐方法
与多组学数据相比,从同一组织的相似但不同细胞中获得每个模态的多模态数据在实验上要容易得多。协调这些数据的任务称为对齐(图1),与另一篇先前综述中描述的对角整合是同义的[17]。应用机器学习和统计方法来完成这项任务的文献非常丰富,包括流形学习、基于神经网络的方法和贝叶斯方法,如表3和表4所总结,并如图3所示。重要的是要注意,在多组学中,我们事先不知道跨组学层的细胞对应关系,因此除了在前一节中描述的学习细胞在多组学整合中的表示的工作之外,还需要额外关注对齐这些表示的分布。因此,专为整合设计的方法通常无法执行对齐。相反,专为对齐设计的方法在整合任务上可能表现不佳。
图3 单细胞多组学对齐方法
展示了对齐多组学单细胞数据的一些常见方法:
- 贝叶斯方法,通过使用多个潜在变量对 -组学测量的概率分布进行建模,并使用贝叶斯公式更新这些分布(A);
- 流形对齐方法,在组学空间中揭示了可以进行对齐的表面(B);
- 以及基于神经网络的模型,创建不同 -组学数据的潜在表示,从而更容易进行对齐(C)。

贝叶斯方法
Clonealign [45]通过将通过RNA-seq测定的细胞分配给由DNA-seq数据衍生的克隆,从而整合了异质群体的单细胞RNA和DNA测序数据。Clonealign基于贝叶斯潜变量模型,其中使用分类变量来指定细胞分配。该模型通过在基因表达上引入拷贝数剂量效应,将基因的拷贝数映射到其表达值。该模型还足够灵活,可以容纳额外的协变量,例如批次效应或可以从基因表达中推断出的生物信息(细胞周期)。除了通过模拟研究展示其稳健性外,Clonealign还应用于真实的癌症数据集,以发现新的克隆特异性失调的生物通路。
在MUSIC [46] 是一种无监督的主题建模方法,用于整合分析单细胞RNA数据和汇聚的簇状规律间隔短回文重复(CRISPR)筛选数据[47]。该模型通过描绘扰动效应来连接细胞的基因表达谱和特定的生物功能,从而更好地理解在单细胞CRISPR数据中的扰动功能。扰动效应优先步骤中,MUSIC利用主题模型的输出,并估计单个基因对细胞表型的扰动效应。它采用了三种不同的方案来建模综合的单细胞和CRISPR数据中的基因扰动效应:整体扰动效应、由主题模型表示的功能主题特异性扰动效应,以及不同扰动效应之间的关系。MUSIC应用于14个真实的单细胞CRISPR筛选数据集,并准确量化和优先考虑了对细胞表型的单个基因扰动效应,对大量噪音具有较高的容忍度。
在这种上下文中,“对齐”指的是将来自不同来源或类型的数据映射到一个共同的框架或空间,以便能够进行比较、分析或集成。
流形对齐方法
流形对齐方法的目标是推断多个复杂数据集中的低维结构(图3B)。一旦完成这一步,就可以在数据集之间匹配数据点。这是一类非常广泛的算法,我们在这里基于不同的思想进行了综述,例如使用伪时间轨迹、核方法以及基于细胞距离的匹配等。
具体来说,对于流形对齐方法,其目标是将不同组学数据(例如,基因表达和甲基化数据)中的特征对准,以便更好地理解它们之间的关系或共同的结构。
MATCHER [48] 是第一种用于对齐不同形式的单细胞数据的流形对齐技术。他们的方法基于轨迹推断[49]。该方法首先为每个组学构建与细胞过程相对应的伪时间轨迹,然后在不同组学之间对齐它们。伪时间轨迹将相应的细胞过程建模为一个高斯过程,并推断与伪时间相对应的潜变量。这产生了一组曲线,每一层组学都对应一条曲线,捕捉了生物过程。然后,这些曲线被投影到参考线上,以便在不同组学之间匹配不同的细胞。该模型假设只有一个共同的生物过程需要建模。
基于最大均值差异的流形对齐(MMD-MA)[50] 是一种完全无监督的方法。对齐是通过匹配不同组学的低维表示来完成的,这些表示是通过一种基于核的技术构建的,该技术通过最小化两个数据集之间的最大均值差异(MMD)[51] 来实现。此外,这些表示是通过考虑原始数据中距离的扭曲来构建的,同时保持变换尽可能简单。该模型在包含来自同一单个细胞的基因表达和甲基化值的数据上进行了评估;已知的细胞对应信息被隐藏,而MMD-MA能够成功地重建这些信息。
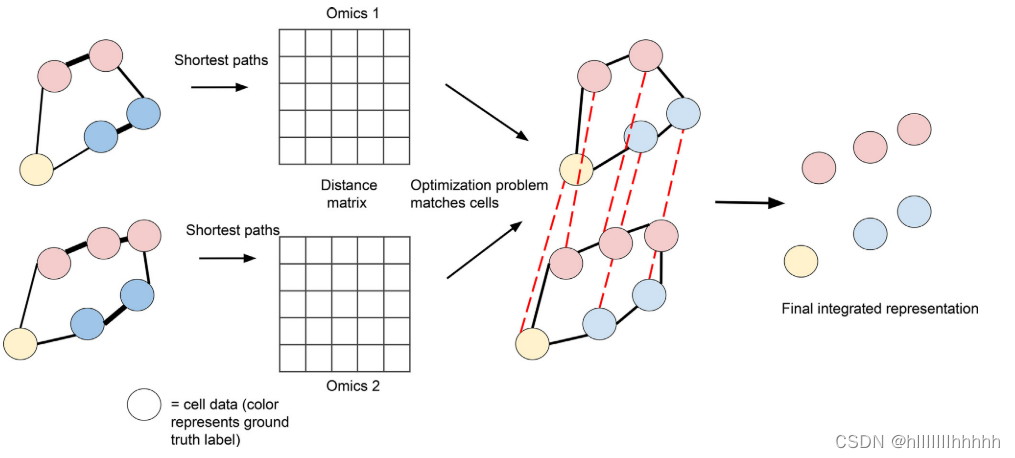
基于距离匹配的方法是一类通过匹配数据集的结构来执行不受监督的不同组学数据对齐的方法(图4)。代表性的方法包括UNION-Com [52]、SCOT [53] 和 Pamona [53]。它们的共同思想是,如果不同的组学层确实对应于相似的细胞样本,则在重新排列细胞索引后,任何两个组学层的距离矩阵将变得非常相似。来自不同组学的细胞之间的距离是通过考虑不同组学层中的kNN图并找到沿图的最近距离来计算的。UNION-Com [52]通过优化细胞排列后的距离矩阵的相似性,找到连接数据集中点的匹配矩阵。这种匹配方法是对广义无监督流形对齐(GUMA)[54]的扩展,引入了新的允许软匹配的概念。随后,该方法执行了一种适用于在相同潜在空间中表示的多模态数据的t-分布随机邻居嵌入(t-SNE)[55]的版本。SCOT [53] 通过最优传输理论执行软匹配,每个最优化问题都有一个不同的优化问题。被最小化的量是Gromov-Wasserstein距离,它将earth-mover Wasserstein距离推广到不同空间之间的最优传输[56]。Pamona [57] 使用了与SCOT类似的方法,但使用基于部分Gromov-Wasserstein距离[58]的最优传输的修改版本,该距离考虑了在数据集之间没有适当匹配的数据点。通过这样做,它允许数据集之间可能存在不完美的对齐,容忍仅存在于一个数据集中的细胞类型。在找到对齐后,与不同模态对应的数据被投影到一个通过Laplacian eigenmaps [59]降维的空间中。这种方法能够在匹配细胞时考虑所有数据集的整体结构,而无需要求不同模态具有相同的分布。
图4 基于距离的对齐
基于距离的对齐算法的示意概述:细胞在两种不同的图表示中由节点表示,分别对应于两种不同的组学测定。具有非常相似组学测量的细胞连接以形成图。然后,为了保持图上的距离概念,对这两个图进行对齐。
在距离匹配方法的原始研究中进行了有限的基准测试。UNION-Com在评估基因表达、甲基化和染色质可及性数据之间标签转移质量时,与Seurat v3和MMD-MA相比表现良好[52]。SCOT在包含转录组和表观遗传(DNA甲基化或染色质可及性)数据的多个真实和模拟数据集上,与MMD-MA和UNION-Com相比表现良好[53]。Pamona在包含转录组和表观遗传数据的多个数据集上,在基准测试中优于SCOT、MMD-MA和Seurat v3 [57]。显然,需要进行更全面的比较,以评估这一类方法在其他建模方法上的性能。
基于神经网络方法
神经网络,包括自编码器和生成对抗网络(GAN),已用于无监督任务中的组学数据集对齐。自编码器早先已有描述。生成对抗网络通常包括两部分:生成器网络和判别器网络。生成器试图产生与某个目标数据集相似的输出形式,而判别器被优化以学习生成器输出与目标数据集元素之间的差异。在本节中,我们总结了下面的相关神经网络方法。
SCIM [60] 基于多领域翻译方法[61],以无监督的方式整合多组学数据。它使用一个独立的变分自编码器为每种模态构建,以将数据映射到降维的潜在空间表示。然后,通过使用鉴别器网络,该网络除了自编码器外,还学习区分不同组学的潜在空间表示,将这些表示对齐以具有相似的结构。两个自编码器和鉴别器网络同时进行训练,使两个潜在空间尽可能相似。一旦两个数据集都被编码成大致对应的表示,具有相似潜在表示的点将在数据集之间匹配。该模型在概率模拟单细胞RNA-seq树状拓扑结构(PROSSTT)[62]以及包含基因表达和蛋白质的数据集的仿真数据上进行了测试,并在应用于展现复杂细胞分化过程的模拟数据时,表现出色于MATCHER。
SCIM(Single-Cell Integration using Multi-domain translation)使用了一种多领域翻译的方法,通过无监督学习将多组学数据进行整合。以下是SCIM对齐的具体实现步骤:
1. **每个模态的变分自编码器:** 对于每个组学模态(例如基因表达、蛋白质表达等),SCIM使用一个独立的变分自编码器。这些自编码器的作用是将原始数据映射到一个降维的潜在空间,以捕捉数据的重要特征。
2. **潜在空间对齐:** 在编码器之后,SCIM引入了一个鉴别器网络。这个鉴别器网络的作用是学习区分不同组学模态的潜在空间表示。通过使用鉴别器网络,SCIM确保了不同模态的潜在表示具有相似的结构。这是通过在训练中优化自编码器和鉴别器网络来实现的。
3. **双模态训练:** SCIM同时训练两个自编码器和鉴别器网络。这意味着在训练期间,它努力使两个组学模态的潜在空间尽可能相似。这样,通过在两个模态上学习,SCIM能够找到它们之间的最佳映射,从而实现对齐。
4. **匹配相似潜在表示:** 一旦两个数据集都被编码成相应的表示,SCIM通过匹配在这两个模态上具有相似潜在表示的点来完成对齐。这确保了在不同组学数据中,具有相似特征的单元被正确地对齐。
在具体的实验中,SCIM在使用概率模拟单细胞RNA-seq数据和包含基因表达和蛋白质表达的真实数据集时进行了测试,并在复杂细胞分化过程的模拟数据上表现优越。SCIM的方法主要基于通过训练自编码器和鉴别器网络来优化潜在空间的结构,从而使不同组学模态的数据能够在降维潜在空间中对齐。
MULTIGRATE [63] 使用多模态变分自编码器结构将多组学数据投影到共享的潜在空间。尽管与scMVAE模型 [28] 有些相似,但这个框架提供了额外的灵活性,并可用于整合成对和非成对的单细胞数据。此外,该模型可以将来自多组学测定(例如CITE-seq)的数据与来自单组学测定(例如scRNA-seq)的数据进行整合。不同组学对应的数据首先通过各自的神经网络传递,然后通过专家模型技术 [29] 进行合并,形成潜在分布。解码器网络然后旨在从这个统一的表示中重建所有组学的数据。为了更好地对齐细胞,MMD(最大均值差异)被添加到损失函数中,惩罚不同测定之间点云的错位。他们的模型被用于创建多模态图谱,并将COVID-19(2019冠状病毒病)单细胞数据集映射到多模态参考上。
MAGAN [64] 利用生成对抗网络(GANs)来对齐来自不同领域的数据。MAGAN使用两个相互关联的GANs在组学层之间进行转换,同时绑定它们的参数,并要求它们的组合将任何点映射到其自身。换句话说,如果第一个生成器将数据点 A 映射到数据点 B,那么第二个生成器应该将 B 映射回 A。在概念上,这与计算机视觉中的CycleGAN [65] 模型非常相似,但具有一个关键创新,使其能够更有效地对齐和整合单细胞数据。创新之处在于注意到,虽然CycleGAN框架在对齐数据集整体上非常出色,但它不一定能够正确匹配个别点。这对于单细胞数据来说是一个特别重要的问题。为了解决这个问题,MAGAN 添加了一个对应损失,用于衡量在生成器映射之前和之后的点之间的差异。该模型在各种数据集上进行了测试,从模拟数据集到修改后的国家标准技术研究所(MNIST)手写数字到分子数据。该方法被应用于合并单细胞中的转录组和蛋白质组数据。即使在没有对应信息的情况下,该模型也被证明能够有意义地对齐数据集。
其他方法
先前开发的一些用于对齐不同的单细胞RNA测序(scRNA-seq)数据集的方法,原则上也可以重新用于单细胞多组学对齐。在这种情况下,不同的组学数据被汇总到基因上,并转换成基因活性得分,与scRNA-seq数据具有相同的格式。在这里,我们介绍了两种这样的方法,LIGER和Seurat,因为它们非常受欢迎。这种方法的一个缺点是无法单独建模组学数据。由于空间限制,我们建议读者参考先前关于其他scRNA-seq整合方法的基准研究[66]。
基于典型相关分析(CCA)的方法通过选择在数据集之间相关的自由度来降低数据的维度。Seurat v3 [67] 结合了CCA和网络概念,以对齐和整合单细胞多组学数据。在执行CCA后,该算法识别数据集之间的锚点并对这些锚点的质量进行评分。锚点是通过相互最近邻(MMN)来识别的,它们的质量是通过考虑锚点邻域之间的重叠来评分的。与Seurat v3类似,MAESTRO [68] 也利用了典型相关分析来整合转录组和表观遗传数据,并提供了全面的分析流程。bindSC [69] 也使用典型相关分析来构建数据的共享表示,通过一个定制的程序进行迭代优化。
LIGER [70] 进行了综合的非负矩阵分解(iNMF)以学习解释数据集内和跨数据集的变异的因子。例如,DNA甲基化等数据首先在基因上进行汇总。对应于不同数据集的细胞由不同的细胞特异性因子集描述。基因因子包括两个组成部分:一个在数据集之间共享的部分,一个是数据集特定的;该模型旨在使数据集特定部分尽量小。在执行矩阵分解后,形成了共享因子邻域图,其中细胞根据其因子的相似性相连接,并用于在模态之间对齐细胞。最近,这种非负矩阵分解方法已经扩展到包括在线学习的概念。它通过实时迭代更新模型,从而实现更好的可扩展性和计算效率 [71]。
相关文章:
单细胞多组学整合与对齐的计算方法
Computational Methods for Single-cell Multi-omics Integration and Alignment Bioinformatics-2022-密西根大学 关键词:单细胞;多组学;机器学习;无监督学习;集成 摘要 最近发展起来的生成单细胞基因组数据的技术在生物学领域产生了革命性的影响。多组学测定提…...

33.openeuler OECA认证模拟题16
一 、选择题 1.如何查看系统支持的 shell? A、cat /etc/passwd B、cat /etc/shells C、echo SSHELL D、echo $0 答案 :B 2.下列哪项不是 shell的功能? A 、 用户界面,提供用户与内核交互接口 B 、 命令解释器 C 、提供编译环境 D 、 提供各种管理工具,…...

javaScript数组去重的几种实现方式——适用非引用数据去重
最传统的使用循环遍历 //最传统的使用循环遍历 function getUnique(arr) {let newArr [];for (let i 0; i < arr.length; i) {for (let j i 1; j < arr.length; j) {if (arr[i] arr[j]) {i; //相同丢掉前面的元素}}newArr.push(arr[i]);}return newArr; } 利用Set实…...
Nexus Repository Manager
Nexus Repository Manager https://s01.oss.sonatype.org/#welcome https://mvnrepository.com/-CSDN博客...

Python世界之运算符
一、算术运算符 以下假设变量: a10,b20: 运算符 描述 实例 加 - 两个对象相加 a b 输出结果 30 - 减 - 得到负数或是一个数减去另一个数 a - b 输出结果 -10 * 乘 - 两个数相乘或是返回一个被重复若干次的字符串 a * b 输出结…...

蓝桥杯倒计时47天!DFS基础——图的遍历
倒计时47天! 深度优先搜索——DFS 温馨提示:学习dfs之前最好先了解一下递归的思想。 DFS基础——图的遍历 仙境诅咒 问题描述 在一片神秘的仙境中,有N位修仙者,他们各自在仙境中独立修炼,拥有自己独特的修炼之道…...
体验LobeChat搭建私人聊天应用
LobeChat是什么 LobeChat 是开源的高性能聊天机器人框架,支持语音合成、多模态、可扩展的(Function Call)插件系统。支持一键免费部署私人 ChatGPT/LLM 网页应用程序。 地址:https://github.com/lobehub/lobe-chat 为什么要用Lobe…...

ClickHouse 指南(三)最佳实践 -- 主键稀疏索引
在ClickHouse主索引的实用介绍 ClickHouse release 24.1, 2024-01-30 1、简介 在本指南中,我们将深入研究ClickHouse索引。我们将详细说明和讨论: ClickHouse中的索引与传统的关系数据库管理系统有何不同ClickHouse是如何构建和使用表的稀疏主索引的什么是在Clic…...

【Nginx】Nginx配置反向代理 和 https
nginx.conf配置 进入linux /etc/nginx/ 打开nginx.conf 进行以下配置 http {include mime.types;default_type application/octet-stream;sendfile on;keepalive_timeout 65;server {#监听443端口listen 443 ssl;#你的域名server_name huiblog.top;#ssl证书的pe…...

ChatGPT第七讲
ChatGPT为什么会被热炒? 2023年上半年,ChatGPT引起了广泛的热议,对于ChatGPT有多热,不需要我重复了,你可能在网上看到了很多报道,标题如《ChatGPT揭开AI战幔:杀死黄页一样摧毁Google?…...
)
Chapter 2 of Effective C++ (构造/析构/赋值运算)
条款06:了解C默默编写并调用哪些函数 Know what functions C silently writes and calls 编译器会为空类生成一个copy构造函数、copy assignment操作符和一个析构函数。此外如果你没有声明任何构造函数,它也会生成一个默认构造函数。 (对C1…...

Android学习笔记 service启动方式
在Android系统中,Service的启动方式主要有两种: ## 1. startService 这种方式用于启动一个服务执行后台任务,不进行通信。当你调用startService()方法启动服务后,服务会一直无限期运行下去,只有在外部调用了stopServi…...
Redis 工具类 与 Redis 布隆过滤器
Redis 工具类 1. 核心依赖 <!--redis--> <dependency><groupId>org.springframework.boot</groupId><artifactId>spring-boot-starter-data-redis</artifactId> </dependency> <dependency><groupId>com.google.guava…...

自定义el-upload 上传文件
前言 最近在做一个文件上传的功能,后端接口写好了、发现前端上传文件的页面不会写……(我很笨的)然后我就找啊找发现element有个组件是<el-upload/>能直接上传文件。我就想直接用拿来改改改成自己想要的,可是就是这样我花了…...
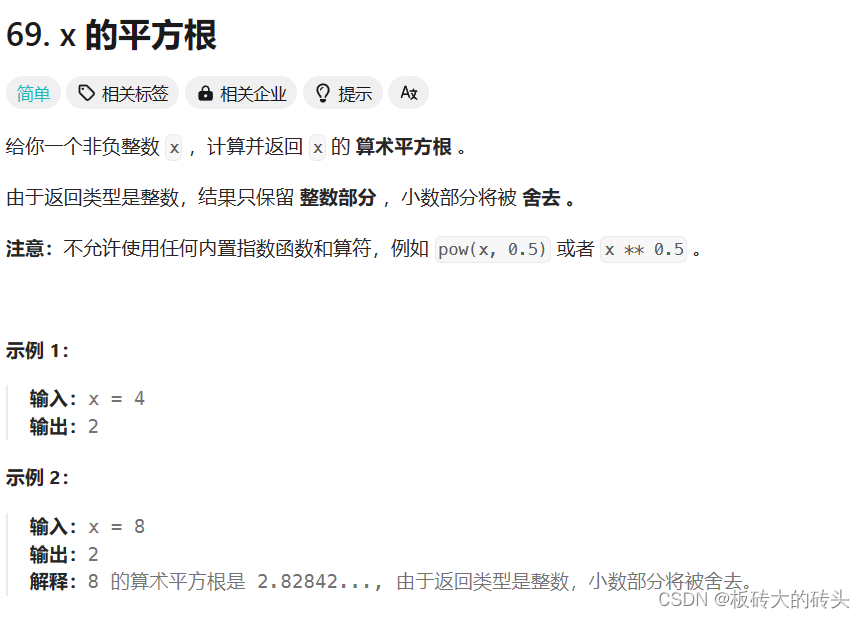
LeetCode69. x 的平方根(C++)
LeetCode69. x 的平方根 题目链接代码 题目链接 https://leetcode.cn/problems/sqrtx/description/ 代码 class Solution { public:int mySqrt(int x) {int right x, left 0, ans -1;while(left < right){long long mid left (right - left) / 2;if(mid * mid <…...

[c++] 单例模式 + cyberrt TimingWheel 单例分析
单例模式要求一个类在一个进程中只能创建一个对象。比如 cyberrt 中的 TimingWheel 类就是单例模式,这个类管理着一个进程内的所有定时器,只需要一个对象就可以。 单例模式的实现有两种方式,懒汉式和饿汉式。懒汉式,当第一次使用…...

如何在cmd里面创建一个vue项目
在命令提示符(CMD)中创建一个Vue项目,你需要先确保你已经全局安装了Vue CLI(Vue的命令行工具)。如果你还没有安装Vue CLI,可以通过以下命令进行安装: bash复制代码 npm install -g vue/cli # O…...

Day2 JS基础
2.1 运算符 赋值运算符 一元运算符 -- <script>let h20let kh hconsole.log(h) //22console.log(k) //42let i1console.log(i i i) //7 // 递增运算符:var a8aconsole.log(a) //9 var num10var bnumconsole.log(b) //10</script> 比较运…...
)
mybatis----有用配置知识归纳(狂神说学习总结)
1.mybatis介绍 MyBatis 是一款优秀的持久层框架MyBatis 避免了几乎所有的 JDBC 代码和手动设置参数以及获取结果集的过程MyBatis 可以使用简单的 XML 或注解来配置和映射原生信息,将接口和 Java 的 实体类映射成数据库中的记录 官网 Mybatis中文官方文档 : https…...

【TCP/IP】组播
一、组播介绍 组播(Multicast)是网络技术中数据传输的一种方法,它允许将数据包同时发送给一组指定的目标,而不是单个的目标(单播 Unicast)或所有可能的目标(广播 Broadcast)。组播传…...

网盘下载新革命:九大平台一键直链,告别客户端束缚
网盘下载新革命:九大平台一键直链,告别客户端束缚 【免费下载链接】Online-disk-direct-link-download-assistant 一个基于 JavaScript 的网盘文件下载地址获取工具。基于【网盘直链下载助手】修改 ,支持 百度网盘 / 阿里云盘 / 中国移动云盘…...

如何快速免费管理游戏DLSS版本?DLSS Swapper终极指南
如何快速免费管理游戏DLSS版本?DLSS Swapper终极指南 【免费下载链接】dlss-swapper 项目地址: https://gitcode.com/GitHub_Trending/dl/dlss-swapper DLSS Swapper是一款革命性的开源工具,专为PC游戏玩家设计,能够智能管理、下载和…...

LrcHelper:3分钟掌握网易云音乐双语歌词下载,告别歌词烦恼
LrcHelper:3分钟掌握网易云音乐双语歌词下载,告别歌词烦恼 【免费下载链接】LrcHelper 从网易云音乐下载带翻译的歌词 Walkman 适配 项目地址: https://gitcode.com/gh_mirrors/lr/LrcHelper 你是否曾为找不到心爱歌曲的歌词而烦恼?或…...

Windows Cleaner终极指南:3步彻底解决C盘爆红问题,让电脑重获新生!
Windows Cleaner终极指南:3步彻底解决C盘爆红问题,让电脑重获新生! 【免费下载链接】WindowsCleaner Windows Cleaner——专治C盘爆红及各种不服! 项目地址: https://gitcode.com/gh_mirrors/wi/WindowsCleaner 还在为Wind…...

3个维度深度解析:UABEA如何重塑Unity资源处理生态
3个维度深度解析:UABEA如何重塑Unity资源处理生态 【免费下载链接】UABEA c# uabe for newer versions of unity 项目地址: https://gitcode.com/gh_mirrors/ua/UABEA 在Unity游戏开发和资源处理的复杂生态中,开发者常常面临一个核心挑战…...

可穿戴电子模块化连接方案:5mm微型按扣实现电路板与织物的可插拔连接
1. 项目概述与核心思路在折腾可穿戴电子项目时,最让人头疼的问题之一,就是如何让电路板与衣物既可靠连接,又能方便地拆下来。传统的做法要么是用导电胶带粘(不牢靠、易氧化),要么是直接把线焊死在板子上然后…...

开源技能图谱工具SkillPort:Go语言构建的知识管理利器
1. 项目概述:一个技能图谱与知识管理的开源利器 最近在整理个人技术栈和团队知识库时,我一直在寻找一个能直观展示技能关联、又能深度管理学习路径的工具。市面上的笔记软件要么太“平”,只能线性记录;要么太“重”,像…...

会话管理封装实践:构建安全可扩展的分布式会话系统
1. 项目概述:一个被低估的会话管理利器如果你是一名开发者,尤其是经常需要处理用户登录、权限校验、状态保持这类“脏活累活”的后端或全栈开发者,那么你一定对“会话管理”这四个字又爱又恨。爱的是,它是构建安全、有状态应用的基…...

Noto Emoji字体架构深度解析:现代表情符号渲染的技术实现与性能优化
Noto Emoji字体架构深度解析:现代表情符号渲染的技术实现与性能优化 【免费下载链接】noto-emoji Noto Emoji fonts 项目地址: https://gitcode.com/gh_mirrors/no/noto-emoji Noto Emoji作为Google开源的表情符号字体库,提供了跨平台的Unicode表…...

终极指南:如何用wxhelper实现PC微信自动化与消息管理
终极指南:如何用wxhelper实现PC微信自动化与消息管理 【免费下载链接】wxhelper Hook WeChat / 微信逆向 项目地址: https://gitcode.com/gh_mirrors/wx/wxhelper wxhelper是一款强大的PC端微信逆向工程工具,通过DLL注入技术为开发者提供完整的微…...