GEO生信数据挖掘(四)数据清洗(离群值处理、低表达基因、归一化、log2处理)
检索到目标数据集后,开始数据挖掘,本文以阿尔兹海默症数据集GSE1297为例
目录
离群值处理
删除 低表达基因
函数归一化,矫正差异
数据标准化—log2处理
完整代码
上节围绕着探针ID和基因名称做了一些清洗工作,还做了重复值检查,空值删除操作。
#查看重复值
table(duplicated(matrix$Gene.Symbol))#去掉缺失值
matrix_na = na.omit(matrix) #基因名称为空删除
matrix_final = matrix_na[matrix_na$Gene.Symbol != "",]离群值处理
用于处理异常值,将超出一定范围的值替换为中位数,以减少异常值对后续分析的影响。
#数据离群处理
#处理极端值
#定义向量极端值处理函数
#用于处理异常值,将超出一定范围的值替换为中位数,以减少异常值对后续分析的影响。
dljdz=function(x) {DOWNB=quantile(x,0.25)-1.5*(quantile(x,0.75)-quantile(x,0.25))UPB=quantile(x,0.75)+1.5*(quantile(x,0.75)-quantile(x,0.25))x[which(x<DOWNB)]=quantile(x,0.5)x[which(x>UPB)]=quantile(x,0.5)return(x)
}#第一列设置为行名
matrix_leave=matrix_finalboxplot(matrix_leave,outline=FALSE, notch=T, las=2) ##出箱线图
dim(matrix_leave)#处理离群值
matrix_leave_res=apply(matrix_leave,2,dljdz)boxplot(matrix_leave_res,outline=FALSE, notch=T, las=2) ##出箱线图
dim(matrix_leave_res)
删除 低表达基因
方案1 :仅去除在所有样本里表达量都为零的基因(或者平均值小于1)
方案2:仅保留在一半(50%,75%...自己选择)以上样本里表达的基因
#删除 低表达基因#仅去除在所有样本里表达量都为零的基因(平均值小于1)# 计算基因表达矩阵的平均值
gene_avg <- apply(matrix_final, 1, mean)
# 根据阈值筛选低表达基因
filtered_genes_1 <- matrix_final[gene_avg >= 1, ] # 表达量平均值小于1的过滤
dim(filtered_genes_1)#+================================
#常用过滤标准2(推荐):
#仅保留在一半以上样本里表达的基因# 计算基因表达矩阵每个基因的平均值
gene_means <- rowMeans(matrix_final)# 计算基因平均值的排序百分位数
gene_percentiles <- rank(gene_means) / length(gene_means)# 获取阈值
threshold <- 0.25 # 删除后25%的阈值
#threshold <- 0.5 # 删除后50%的阈值
# 根据阈值筛选低表达基因
filtered_genes_2 <- matrix_final[gene_percentiles > threshold, ]# 打印筛选后的基因表达矩阵
dim(filtered_genes_2)boxplot(filtered_genes_2,outline=FALSE, notch=T, las=2) ##出箱线图
#dim(filtered_genes)#+删除 低表达基因 得到filtered_genes_1 删除平均表达小于1的 得到filtered_genes_2 删除后25%的函数归一化,矫正差异
#1.归一化不是绝对必要的,但是推荐进行归一化。
#有重复的样本中,应该不具备生物学意义的外部因素会影响单个样品的表达,
#例如中第一批制备的样品会总体上表达高于第二批制备的样品,假设所有样品表达值的范围和分布都应当相似,
#需要进行归一化来确保整个实验中每个样本的表达分布都相似。
#2.归一化要在log2标准化之前做
library(limma) exprSet=normalizeBetweenArrays(filtered_genes_2)boxplot(exprSet,outline=FALSE, notch=T, las=2) ##出箱线图## 这步把矩阵转换为数据框很重要
class(exprSet) ##注释:此时数据的格式是矩阵(Matrix)
exprSet <- as.data.frame(exprSet)数据标准化—log2处理
如果表达量的数值在50以内,通常是经过log2转化后的。如果数字在几百几千,则是未经转化的。
GSE数据集的注释部分会有说明,常见的标准化处理方法有3种:RMA算法、GC-RMA算法、MAS5算法
#标准化 表达矩阵自动log2化
qx <- as.numeric(quantile(exprSet, c(0., 0.25, 0.5, 0.75, 0.99, 1.0), na.rm=T))
LogC <- (qx[5] > 100) ||(qx[6]-qx[1] > 50 && qx[2] > 0) ||(qx[2] > 0 && qx[2] < 1 && qx[4] > 1 && qx[4] < 2)## 开始判断
if (LogC) { exprSet [which(exprSet <= 0)] <- NaN## 取log2exprSet_clean <- log2(exprSet)print("log2 transform finished")
}else{print("log2 transform not needed")
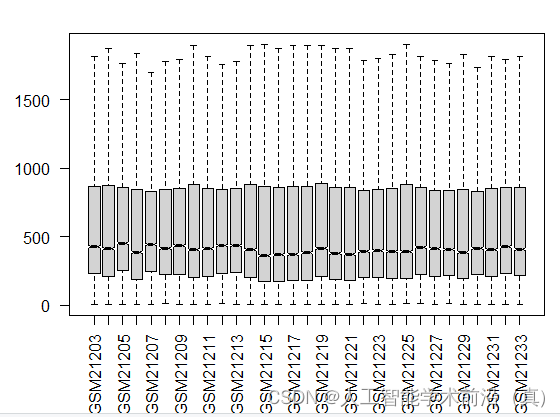
}筛选后的基因表达矩阵的箱线图
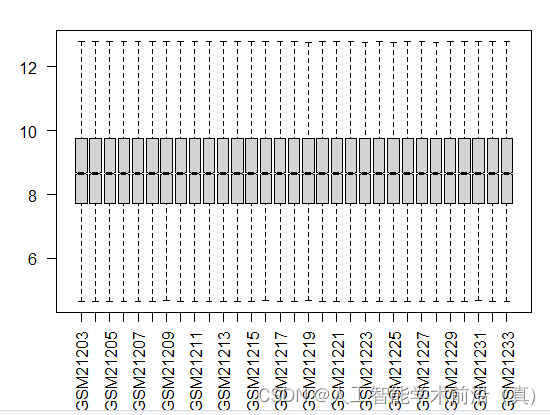
经过数据清洗后的箱线图
完整代码
# 安装并加载GEOquery包
library(GEOquery)# 指定GEO数据集的ID
gse_id <- "GSE1297"# 使用getGEO函数获取数据集的基础信息
gse_info <- getGEO(gse_id, destdir = ".", AnnotGPL = F ,getGPL = F) # Failed to download ./GPL96.soft.gz!# 提取基因表达矩阵
expression_data <- exprs(gse_info[[1]])# 提取注释信息
#annotation <- featureData(gse_info[[1]])#查看平台文件列名
colnames(annotation)#打印项目文件列表
dir() # 读取芯片平台文件txt
platform_file <- read.delim("GPL96-57554.txt", header = TRUE, sep = "\t", comment.char = "#")#查看平台文件列名
colnames(platform_file)# 假设芯片平台文件中有两列,一列是探针ID,一列是基因名
#probe_names <- platform_file$ID
#gene_symbols <- platform_file$Gene.Symbol
platform_file_set=platform_file[,c(1,11)]#一个探针对应多个基因名,保留第一个基因名
ids = platform_file_set
library(tidyverse)
test_function <- apply(ids,1,function(x){paste(x[1],str_split(x[2],'///',simplify=T),sep = "...")})
x = tibble(unlist(test_function))colnames(x) <- "ttt"
ids <- separate(x,ttt,c("ID","Gene.Symbol"),sep = "\\...")
dim(ids)#将Matrix格式表达矩阵转换为data.frame格式
exprSet <- data.frame(expression_data)
dim(exprSet)#给表达矩阵新增加一列ID
exprSet$ID <- rownames(exprSet) # 得到表达矩阵,行名为ID,需要转换,新增一列
dim(exprSet)
#矩阵表达文件和平台文件有相同列‘ID’,使用merge函数合并
express <- merge(x = exprSet, y = ids, by.x = "ID")#删除探针ID列
express$ID =NULLdim(express) matrix = express
dim(matrix)
#查看多少个基因重复了
table(duplicated(matrix$Gene.Symbol))#把重复的Symbol取平均值
matrix <- aggregate(.~Gene.Symbol, matrix, mean) ##把重复的Symbol取平均值
row.names(matrix) <- matrix$Gene.Symbol #把行名命名为SYMBOLdim(matrix)matrix_na = na.omit(matrix) #去掉缺失值
dim(matrix_na)matrix_final = matrix_na[matrix_na$Gene.Symbol != "",]
dim(matrix_final)matrix_final <- subset(matrix_final, select = -1) #删除Symbol列(一般是第一列)
dim(matrix_final)
#+ 经过注释、探针名基因名处理、删除基因名为空值、删除缺失值 得到最终 matrix_final
#+==================================================================================
#+========================================================================================#+==================================================================================
#+========================================================================================
#+增加#(2)离群值处理#数据离群处理
#处理极端值
#定义向量极端值处理函数
#用于处理异常值,将超出一定范围的值替换为中位数,以减少异常值对后续分析的影响。
dljdz=function(x) {DOWNB=quantile(x,0.25)-1.5*(quantile(x,0.75)-quantile(x,0.25))UPB=quantile(x,0.75)+1.5*(quantile(x,0.75)-quantile(x,0.25))x[which(x<DOWNB)]=quantile(x,0.5)x[which(x>UPB)]=quantile(x,0.5)return(x)
}#第一列设置为行名
matrix_leave=matrix_finalboxplot(matrix_leave,outline=FALSE, notch=T, las=2) ##出箱线图
dim(matrix_leave)#处理离群值
matrix_leave_res=apply(matrix_leave,2,dljdz)boxplot(matrix_leave_res,outline=FALSE, notch=T, las=2) ##出箱线图
dim(matrix_leave_res)#+增加#(2)离群值处理
#+==================================================================================
#+========================================================================================#+==================================================================================
#+========================================================================================
#删除 低表达基因#仅去除在所有样本里表达量都为零的基因(平均值小于1)# 计算基因表达矩阵的平均值
gene_avg <- apply(matrix_final, 1, mean)
# 根据阈值筛选低表达基因
filtered_genes_1 <- matrix_final[gene_avg >= 1, ] # 表达量平均值小于1的过滤
dim(filtered_genes_1)#+================================
#常用过滤标准2(推荐):
#仅保留在一半以上样本里表达的基因# 计算基因表达矩阵每个基因的平均值
gene_means <- rowMeans(matrix_final)# 计算基因平均值的排序百分位数
gene_percentiles <- rank(gene_means) / length(gene_means)# 获取阈值
threshold <- 0.25 # 删除后25%的阈值
#threshold <- 0.5 # 删除后50%的阈值
# 根据阈值筛选低表达基因
filtered_genes_2 <- matrix_final[gene_percentiles > threshold, ]# 打印筛选后的基因表达矩阵
dim(filtered_genes_2)boxplot(filtered_genes_2,outline=FALSE, notch=T, las=2) ##出箱线图
#dim(filtered_genes)#+删除 低表达基因 得到filtered_genes_1 删除平均表达小于1的 得到filtered_genes_2 删除后25%的
#+==================================================================================
#+========================================================================================#+==================================================================================
#+========================================================================================
### limma的函数归一化,矫正差异 ,表达矩阵自动log2化#1.归一化不是绝对必要的,但是推荐进行归一化。
#有重复的样本中,应该不具备生物学意义的外部因素会影响单个样品的表达,
#例如中第一批制备的样品会总体上表达高于第二批制备的样品,假设所有样品表达值的范围和分布都应当相似,
#需要进行归一化来确保整个实验中每个样本的表达分布都相似。
#2.归一化要在log2标准化之前做library(limma) exprSet=normalizeBetweenArrays(filtered_genes_2)boxplot(exprSet,outline=FALSE, notch=T, las=2) ##出箱线图## 这步把矩阵转换为数据框很重要
class(exprSet) ##注释:此时数据的格式是矩阵(Matrix)
exprSet <- as.data.frame(exprSet)#标准化 表达矩阵自动log2化
qx <- as.numeric(quantile(exprSet, c(0., 0.25, 0.5, 0.75, 0.99, 1.0), na.rm=T))
LogC <- (qx[5] > 100) ||(qx[6]-qx[1] > 50 && qx[2] > 0) ||(qx[2] > 0 && qx[2] < 1 && qx[4] > 1 && qx[4] < 2)## 开始判断
if (LogC) { exprSet [which(exprSet <= 0)] <- NaN## 取log2exprSet_clean <- log2(exprSet)print("log2 transform finished")
}else{print("log2 transform not needed")
}boxplot(exprSet_clean,outline=FALSE, notch=T, las=2) ##出箱线图
### limma的函数归一化,矫正差异 ,表达矩阵自动log2化 得到exprSet_clean
#+==================================================================================
#+========================================================================================# saving all data to the path
save(exprSet_clean, file ="exprSet_clean_75percent_filter.RData")上述处理得到了干净的基因表达矩阵,数据部分已经没有问题,但是在做数据挖掘(差异分析、富集分析等)之前还有一项准备工作,要将数据样本进行分组,即患病组、对照组。
相关文章:
GEO生信数据挖掘(四)数据清洗(离群值处理、低表达基因、归一化、log2处理)
检索到目标数据集后,开始数据挖掘,本文以阿尔兹海默症数据集GSE1297为例 目录 离群值处理 删除 低表达基因 函数归一化,矫正差异 数据标准化—log2处理 完整代码 上节围绕着探针ID和基因名称做了一些清洗工作,还做了重复值检查…...

CI/CD工具中的CI和CD的含义
CI/CD工具中的CI和CD的含义? CI/CD 是现代软件开发方法中广泛使用的一种方法。其中,CI 代表持续集成(Continuous Integration),CD 则有两层含义,一是持续交付(Continuous Delivery)…...

用go获取IPv4地址,WLAN的IPv4地址,本机公网IP地址详解
文章目录 获取IPv4地址获取WLAN的IPv4地址获取本机公网IP地址 获取IPv4地址 下面的代码会打印出本机所有的IPv4地址。这个方法可能会返回多个IP地址,因为一台机器可能有多个网络接口,每个接口可能有一个或多个IP地址。 package mainimport ("fmt&…...

Android自定义Drawable---灵活多变的矩形背景
Android自定义Drawable—灵活多变的矩形背景 在安卓开发中,我们通常需要为不同的按钮设置不同的背景以实现不同的效果,有时还需要这些按钮根据实际情况进行变化。如果采用编写resource中xml文件的形式,就需要重复定义许多只有微小变动的资源…...

ParagonNTFSforMac_15.5.102中文版最受欢迎的NTFS硬盘格式读取工具
Paragon NTFS for Mac是一款可以为您轻松解决Mac平台上不能识别Windows通用的NTFS文件难题,这是一款强大的Mac读写工具,相信在很多时候,Mac用户需要对NTFS文件的移动硬盘进行写入,但是macOS系统默认是不让写入的,使用小…...

Kafka 搭建过程
目录 1.关于Kafka2.Kafka 搭建过程3.参考 本文主要介绍Kafka基本原理,以及搭建过程。 1.关于Kafka Apache Kafka是一个开源的分布式事件流平台,被设计用来实现实时数据流的发布、订阅、存储和处理。 Kafka的主要特性包括: 高吞吐量&#x…...
七、2023.10.1.Linux(一).7
文章目录 1、 Linux中查看进程运行状态的指令、查看内存使用情况的指令、tar解压文件的参数。2、文件权限怎么修改?3、说说常用的Linux命令?4、说说如何以root权限运行某个程序?5、 说说软链接和硬链接的区别?6、说说静态库和动态…...
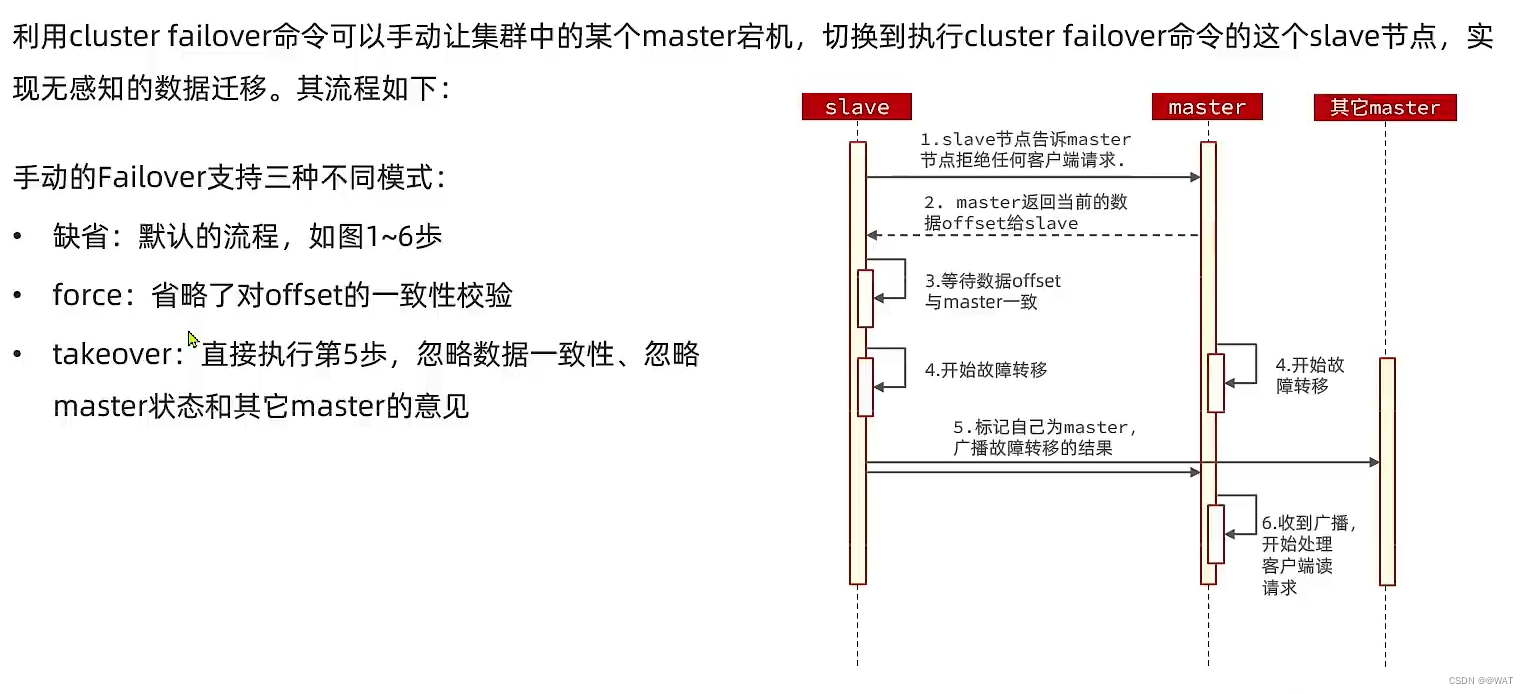
一文教你搞懂Redis集群
一、Redis主从 1.1、搭建主从架构 单节点的Redis的并发能力是有上限的,要进一步的提高Redis的并发能力,据需要大家主从集群,实现读写分离。 共包含三个实例,由于资源有限,所以在一台虚拟机上,开启多个red…...

树上启发式合并 待补
对于每个子树,直接遍历所有轻儿子,继承重儿子 会了板子后,修改维护的东西和莫队是一样的 洛谷 U41492 #include <bits/stdc.h> #define ll long long #define ull unsigned long long constexpr int N1e55; std::vector<int> e…...

minio分布式文件存储
基本介绍 什么是 MinIO MinIO 是一款基于 Go 语言的高性能、可扩展、云原生支持、操作简单、开源的分布式对象存储产品。基于 Apache License v2.0 开源协议,虽然轻量,却拥有着不错的性能。它兼容亚马逊S3云存储服务接口。可以很简单的和其他应…...

Linux新的IO模型io_uring
一、Linux下的网络通信模型 在网络开发的过程中,需要处理好几个问题。首先是通信的内核支持问题;其次是通信的模型问题;最后是框架问题。这些问题在闭源的OS如Windows上,基本上不算什么大问题(因为只能用人家的API&am…...
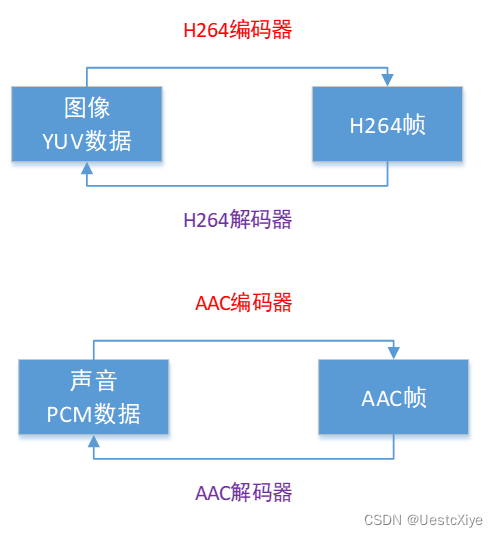
FFmpeg 命令:从入门到精通 | FFmpeg 基本介绍
FFmpeg 命令:从入门到精通 | FFmpeg 基本介绍 FFmpeg 命令:从入门到精通 | FFmpeg 基本介绍FFmpeg 简介FFmpeg 基础知识复用与解复用编解码器码率和帧率 资料 FFmpeg 命令:从入门到精通 | FFmpeg 基本介绍 本系列文章要解决的问题࿱…...

数组篇 第一题:删除排序数组中的重复项
更多精彩内容请关注微信公众号:听潮庭。 第一题:删除排序数组中的重复项 给你一个 非严格递增排列 的数组 nums ,请你 原地 删除重复出现的元素,使每个元素 只出现一次 ,返回删除后数组的新长度。元素的 相对顺序 应…...
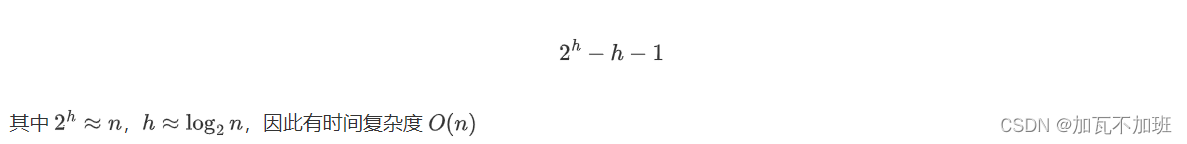
堆的初步认识
在学习本节文章前要先了解:大顶堆与小顶堆: (优先级队列_加瓦不加班的博客-CSDN博客) 堆实现 计算机科学中,堆是一种基于树的数据结构,通常用完全二叉树实现。 什么叫完全二叉树? 答&#x…...

CycleGAN模型之Pytorch实战
一、CycleGAN基本介绍 1. CycleGAN论文:《Unpaired Image-to-Image Translation using Cycle-Consistent Adversarial Networks》 2. 原文代码:https://github.com/junyanz/pytorch-CycleGAN-and-pix2pix 3. 网传精简代码:https://github.com/aitorzip/PyTorch-CycleGAN …...
C++(STL容器适配器)
前言: 适配器也称配接器(adapters)在STL组件的灵活组合运用功能上,扮演着轴承、转换器的角色。 《Design Patterns》对adapter的定义如下:将一个class的接口转换为另一个class的接口,使原本因接口不兼容而…...
)
软考 系统架构设计师系列知识点之软件架构风格(7)
接前一篇文章:软考 系统架构设计师系列知识点之软件架构风格(6) 这个十一注定是一个不能放松、保持“紧”的十一。由于报名了全国计算机技术与软件专业技术资格(水平)考试,11月4号就要考试,因此…...

【Vue3】自定义指令
除了 Vue 内置的一系列指令 (比如 v-model 或 v-show) 之外,Vue 还允许你注册自定义的指令 (Custom Directives)。 1. 生命周期钩子函数 一个自定义指令由一个包含类似组件生命周期钩子的对象来定义。钩子函数会接收到指令所绑定元素作为其参数。 在 <script …...

UG\NX CAM二次开发 加工模块获取 UF _ask_application_module
文章作者:代工 来源网站:NX CAM二次开发专栏 简介: UG\NX CAM二次开发 加工模块获取 UF _ask_application_module 代码: void MyClass::do_it() { // TODO: add your code here // 获取NX当前所在的模块 int module_id = 0; // UF_ask_application_module(&…...

借助GPU算力编译Android
借助GPU算力编译Android 借助GPU编译Android代码的意义在于提高编译的效率和速度。传统的CPU编译方式在处理大量代码时可能会遇到性能瓶颈,而GPU编译利用了显卡的并行计算能力,可以同时处理多个任务,加快编译过程。通过利用GPU的并行计算能力,可以将编译过程中的多个任务分…...

Auto-Coder使用GPT-4o完成:在用TabPFN这个模型构建一个预测未来3天涨跌的分类任务
通过akshare库,获取股票数据,并生成TabPFN这个模型 可以识别、处理的格式,写一个完整的预处理示例,并构建一个预测未来 3 天股价涨跌的分类任务 用TabPFN这个模型构建一个预测未来 3 天股价涨跌的分类任务,进行预测并输…...

涂鸦T5AI手搓语音、emoji、otto机器人从入门到实战
“🤖手搓TuyaAI语音指令 😍秒变表情包大师,让萌系Otto机器人🔥玩出智能新花样!开整!” 🤖 Otto机器人 → 直接点明主体 手搓TuyaAI语音 → 强调 自主编程/自定义 语音控制(TuyaAI…...

【OSG学习笔记】Day 16: 骨骼动画与蒙皮(osgAnimation)
骨骼动画基础 骨骼动画是 3D 计算机图形中常用的技术,它通过以下两个主要组件实现角色动画。 骨骼系统 (Skeleton):由层级结构的骨头组成,类似于人体骨骼蒙皮 (Mesh Skinning):将模型网格顶点绑定到骨骼上,使骨骼移动…...

Unit 1 深度强化学习简介
Deep RL Course ——Unit 1 Introduction 从理论和实践层面深入学习深度强化学习。学会使用知名的深度强化学习库,例如 Stable Baselines3、RL Baselines3 Zoo、Sample Factory 和 CleanRL。在独特的环境中训练智能体,比如 SnowballFight、Huggy the Do…...
SpringCloudGateway 自定义局部过滤器
场景: 将所有请求转化为同一路径请求(方便穿网配置)在请求头内标识原来路径,然后在将请求分发给不同服务 AllToOneGatewayFilterFactory import lombok.Getter; import lombok.Setter; import lombok.extern.slf4j.Slf4j; impor…...
Map相关知识
数据结构 二叉树 二叉树,顾名思义,每个节点最多有两个“叉”,也就是两个子节点,分别是左子 节点和右子节点。不过,二叉树并不要求每个节点都有两个子节点,有的节点只 有左子节点,有的节点只有…...

基于matlab策略迭代和值迭代法的动态规划
经典的基于策略迭代和值迭代法的动态规划matlab代码,实现机器人的最优运输 Dynamic-Programming-master/Environment.pdf , 104724 Dynamic-Programming-master/README.md , 506 Dynamic-Programming-master/generalizedPolicyIteration.m , 1970 Dynamic-Programm…...

CSS设置元素的宽度根据其内容自动调整
width: fit-content 是 CSS 中的一个属性值,用于设置元素的宽度根据其内容自动调整,确保宽度刚好容纳内容而不会超出。 效果对比 默认情况(width: auto): 块级元素(如 <div>)会占满父容器…...
混合(Blending))
C++.OpenGL (20/64)混合(Blending)
混合(Blending) 透明效果核心原理 #mermaid-svg-SWG0UzVfJms7Sm3e {font-family:"trebuchet ms",verdana,arial,sans-serif;font-size:16px;fill:#333;}#mermaid-svg-SWG0UzVfJms7Sm3e .error-icon{fill:#552222;}#mermaid-svg-SWG0UzVfJms7Sm3e .error-text{fill…...
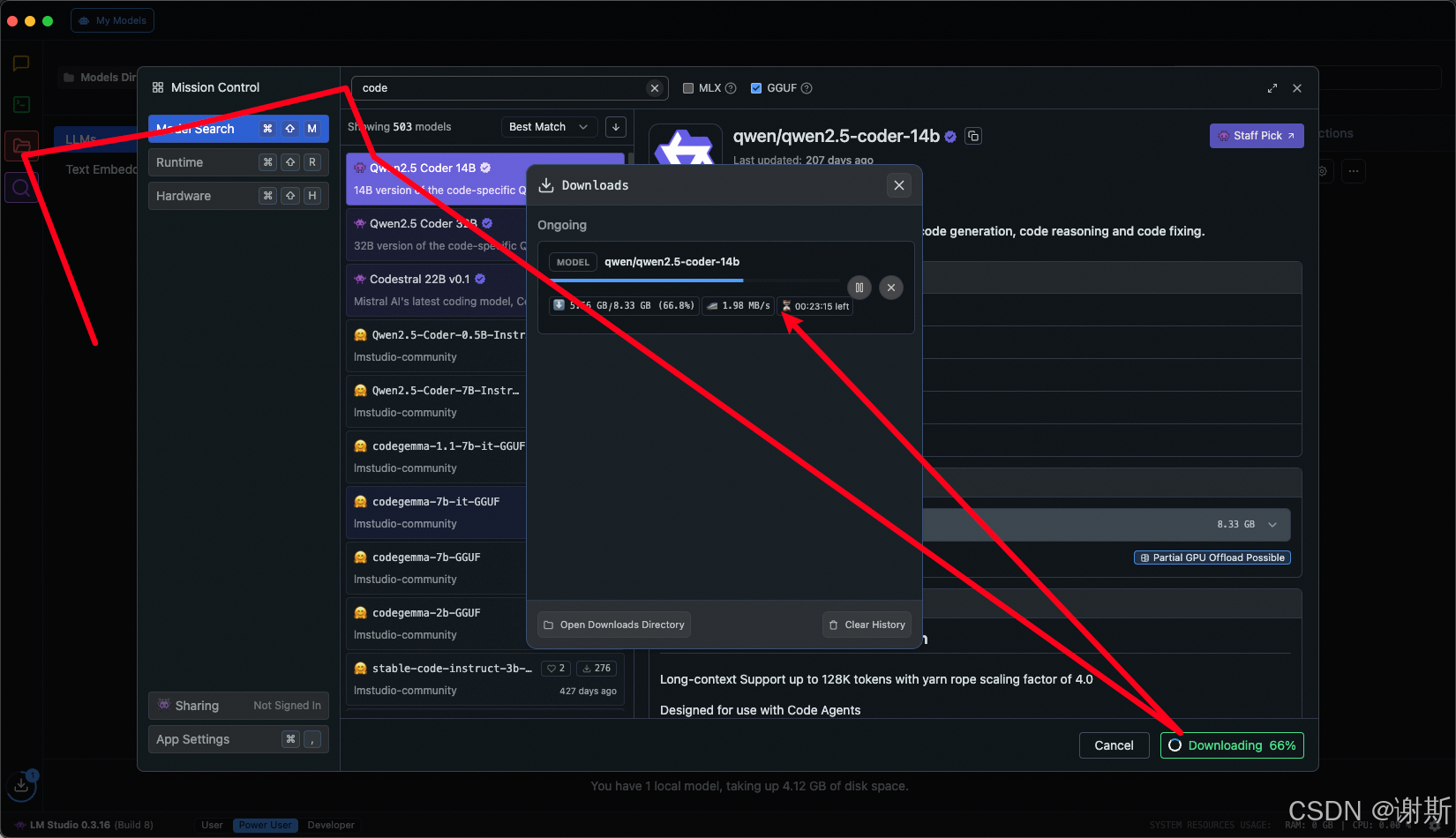
[大语言模型]在个人电脑上部署ollama 并进行管理,最后配置AI程序开发助手.
ollama官网: 下载 https://ollama.com/ 安装 查看可以使用的模型 https://ollama.com/search 例如 https://ollama.com/library/deepseek-r1/tags # deepseek-r1:7bollama pull deepseek-r1:7b改token数量为409622 16384 ollama命令说明 ollama serve #:…...