【2023年中国高校大数据挑战赛 】赛题 B DNA 存储中的序列聚类与比对 Python实现
【2023年中国高校大数据挑战赛 】赛题 B DNA 存储中的序列聚类与比对 Python实现
更新时间:2023-12-29
1 题目
赛题 B DNA 存储中的序列聚类与比对
近年来,随着新互联网设备的大量涌入和对其服务需求的指数级增长,越来越多的数据信息被产生与收集。预计到 2021 年,数据中心内部的IP流量将达到14.7 ZB,数据中心之间的流量将达到 2.8 ZB。如何储存与运输如此庞大的数据已经成为了难题。DNA存储技术是一项着眼于未来的具有划时代意义存储技术,正成为应对数据爆炸的关键技术之一。DNA存储技术指的是使用人工合成的脱氧核糖核苷酸(DNA)作为介质进行信息存储的技术,其具有理论存储量大、维护方便的优点。具体来说,DNA存储将计算机的二进制信息转换为四种碱基(腺嘌呤A、胸腺嘧啶T、鸟嘌呤G和胞嘧啶C)组成的DNA序列(相当于转换为四进制),之后合成为DNA分子干粉。需要读取信息时,将DNA分子进行PCR扩增(这步将会使得原有DNA序列进行扩增复制),之后使用测序仪测出DNA信息。然而在合成、测序等阶段会存在一定的错误,有概率随机发生碱基删除、增添或者替换。下图是某个序列合成测序后的示意图,可以看出由于发生了碱基删除、增添和替换,进而将ATGCATGC变成了AGCAATTC:

因此,对于我们设计好的DNA序列,实际生产测序出来后的序列会存在以下差异:
-
测序后的序列将比原始序列的数量多很多,因为原始序列会被随机扩增成很多条。
-
测序后的序列相比于原始序列有可能存在错误,包括某个碱基缺失、替换、或添加了某个未知碱基,甚至会出现断链。
针对以上两个特点,目前往往需要对测序后的序列进行聚类与比对。其中聚类指的是将测序序列聚类以判断原始序列有多少条,聚类后相同类的序列定义为一个簇。比对则是指在聚类基础上对一个簇内的序列进行比对进而输出一条最有可能的正确序列。通过聚类与比对将会极大地恢复原始序列的信息,但需要注意由于DNA测序后序列众多,如何高效地进行聚类与比对则是在满足准确率基础上的另一大难点。
“train_reference.txt”是某次合成的目标序列,其中第一行为序号,第二行为序列内容。通过真实合成、测序后读取到的测序序列文件为“train_reads.txt”,我们已经对测序序列进行了分类,该文件第一行为目标序列的序号,第二行为序列内容。
基于赛题提供的数据,自主查阅资料,选择合适的方法完成如下任务:
**任务 1:**观察数据集“train_reads.txt”、“train_reference.txt”,针对这次合成任务,进行错误率(插入、删除、替换、断链)、拷贝数方面的分析。其中错误率定义为某个碱基发生错误的概率,需要对不同类型的错误率分别进行分析。拷贝数定义为原始序列复制的数量。
**任务 2:**设计开发一种模型用于对测序后的序列“train_reads.txt”进行聚类,并根据“train_reads.txt”的标签验证模型准确性。模型主要从两方面评估效果:
(1)聚类后准确性(包括簇的数量以及簇内纯度)、(2)聚类速度(以分钟为单位)。
任务 3: “test_reads.txt”是我们在另一种合成环境下合成的测序文件(与 “train_reads.txt”的目标序列不相同),请用任务 2 所开发的模型对其进行聚类,给出聚类耗时以及“test_reads.txt”的目标序列数量,给出拷贝数分布图。
任务 4: 聚类后能否通过比对恢复原始信息也是极为关键的,设计开发一种用于同簇序列的比对模型,该模型可以针对同簇的DNA序列进行比对并输出最有可能正确的目标序列。 请使用该工具对任务 3 中“test_reads.txt”的聚类后序列进行比对,并输出“test_reads.txt”最有可能的目标序列,并分析“test_reads.txt”的错误率。(请用一个“test_ref.txt”的文件记录“test_reads.txt”的目标序列,文件内序列的形式为:
AAAA……
AAAT……
AATA……
……
CCCC……
即序列只用回车间隔,不需要加其他符号,序列顺序按照从前到后,ATGC依次的顺序。此外,需要在论文中展示前十条目标序列的聚类结果。)
附件 1:train_reference.txt train数据集的正确序列
附件 2:train_reads.txt train数据集的合成测序后序列
附件 3:test_reads.txt test数据集的合成测序后序列
参考文献:
-
Dong Y, Sun F, Ping Z, et al. DNA storage: research landscape and future prospects[J]. National Science Review, 2020, 7(6): 1092-1107.
-
Fu L, Niu B, Zhu Z, et al. CD-HIT: accelerated for clustering the next-generation sequencing data[J]. Bioinformatics, 2012, 28(23): 3150-3152.
2 问题分析
2.1 问题一
定义一个函数来比较两个字符串序列,可以自己写for循环去比较,也可以使用字符串比较工具SequenceMatcher。
2.2 问题二
DNA序列的聚类可以采用基于字符串相似度的聚类方法,比如Levenshtein、SMITH-WATERMAN、N-gram方法、或基于序列编码(如k-mer计数)的机器学习聚类方法。
2.3 问题三
在问题二的基础上,对train_reads.xt和test_reads文件和k-mer词频矩阵进行聚类分析,以判断原始序列有多少条。统计每个簇中的序列数量,得到拷贝数分布图。
2.4 问题四
(1)同簇的DNA序列比对方法:对每个簇中的序列进行多数投票,多数序列中出现的碱基将被选为最终序列的对应位置的碱基.
(2)对于每个聚类簇,进行列方向的比对,也就是对于序列的每个位置,从属于该簇的所有序列中选取每个位置上最常出现的碱基作为该位置的最终碱基。
(3)对多数投票的结果,进一步进行相似性评分,比较每个簇的共识序列(从投票中获得的序列)与引用序列库(理想的序列)中的序列。
(4)对于找到的共识序列,将其结果按照聚类簇的索引排序并输出,以方便与目标序列文件(“test_ref.txt”)进行比对,来确定错误位置和错误率。
(5)改进角度:使用更加复杂的比对算法,例如全局比对、局部比对算法、Smith-Waterman、Needleman-Wunsch算法,这些算法考虑了插入、删除和替换,并能够为每种类型的差错提供权重。
3 Python实现
3.1 问题一
import pandas as pd
from difflib import SequenceMatcher
from collections import Counter
from pyecharts.charts import Bar, Pie
from pyecharts import options as opts# 读取目标序列文件和测序序列文件
reference_seq_s = pd.read_csv('data/train_reference.txt',sep=' ',names=['ID','DNA_ref'])
reads = pd.read_csv('data/train_reads.txt',sep=' ',names=['ID','DNA'])
merged_df = pd.merge(reference_seq_s, reads, on='ID', how='inner')# 初始化统计变量
insertion_errors = 0
deletion_errors = 0
replacement_errors = 0
chain_breaks = 0
copy_numbers = Counter()# 定义一个函数来比较两个序列,并统计不同类型的错误
def analyze_sequence(ref_seq, test_seq):global insertion_errors, deletion_errors, replacement_errors, chain_breaks# 略for tag, i1, i2, j1, j2 in s.get_opcodes():if tag == 'replace':replacement_errors += max(i2 - i1, j2 - j1)elif tag == 'delete':deletion_errors += (i2 - i1)elif tag == 'insert':insertion_errors += (j2 - j1)elif tag == 'equal':pass # No errorif len(ref_seq) != len(test_seq):chain_breaks += 1# 进行错误统计和拷贝数计算
for index, row in merged_df.iterrows():analyze_sequence(row['DNA_ref'], row['DNA'])copy_numbers[row['ID']] += 1
# 总的测序次数
total_reads = len(merged_df)# 绘制错误率和拷贝数统计图
def create_charts():# 错误率统计图error_bar = (Bar(init_opts=opts.InitOpts(width="700px", height="500px")).add_xaxis(['Insertion', 'Deletion', 'Replacement', 'Chain Breaks']).add_yaxis('Errors', [insertion_errors, deletion_errors, replacement_errors, chain_breaks]).set_global_opts(title_opts=opts.TitleOpts(title="DNA Sequence Errors")))# 拷贝数统计图copy_num_pie = (Pie(init_opts=opts.InitOpts(width="700px", height="500px")).add("",[list(z) for z in zip([str(id) for id in copy_numbers.keys()], copy_numbers.values())],radius=["40%", "75%"],).set_global_opts(title_opts=opts.TitleOpts(title="DNA Sequence Copy Numbers"),legend_opts=opts.LegendOpts(orient="vertical", pos_top="15%", pos_left="2%"),).set_series_opts(label_opts=opts.LabelOpts(formatter="{b}: {c}")))return error_bar, copy_num_pie# 创建和渲染图表
error_bar, copy_num_pie = create_charts()
error_bar.render("breakdown_of_errors.html")
copy_num_pie.render("dna_copy_numbers.html")

3.2 问题二
方法一:基于Levenshtein距离的聚类算法
import pandas as pd
from sklearn.cluster import AgglomerativeClustering
import Levenshtein
import time# 读取数据
reference_seq_s = pd.read_csv('data/train_reference.txt', sep=' ', names=['ID', 'DNA_ref'])
reads = pd.read_csv('data/train_reads.txt', sep=' ', names=['ID', 'DNA'])# 计算Levenshtein距离矩阵(由于计算量大,这里只示范计算前n个序列的距离矩阵)
n = len(reads)
distance_matrix = [[0] * n for _ in range(n)]
for i in range(n):for j in range(i+1, n):略。。。# 聚类
start_time = time.time()
clustering_model = AgglomerativeClustering(affinity='precomputed', linkage='complete', n_clusters=None, distance_threshold=1.0)
clustering_model.fit(distance_matrix)
duration = time.time() - start_time# 评估聚类结果,这里计算不同簇的数量
clusters = clustering_model.labels_
cluster_counts = pd.Series(clusters).value_counts()import numpy as np
import matplotlib.pyplot as plt
from scipy.cluster.hierarchy import dendrogram# 画出树状图
def plot_dendrogram(model, **kwargs):children = model.children_distance = np.arange(children.shape[0])no_of_observations = np.arange(2, children.shape[0]+2)linkage_matrix = np.column_stack([children, distance, no_of_observations]).astype(float)dendrogram(linkage_matrix, **kwargs)plt.figure(figsize=(15, 8))
plot_dendrogram(clustering_model, labels=range(len(reads)))
plt.ylabel("Distance")
plt.savefig('img/层次聚类.png',dpi=100)
plt.show()
方法二:基于SMITH-WATERMAN算法的聚类
import pandas as pd
import numpy as np
from sklearn.cluster import AgglomerativeClustering
import matplotlib.pyplot as plt
import itertools
# from Bio import pairwise2# 数据读取
reference_seq_s = pd.read_csv('data/train_reference.txt', sep=' ', names=['ID','DNA_ref'])
reads = pd.read_csv('data/train_reads.txt', sep=' ', names=['ID','DNA'])# SMITH-WATERMAN算法的实现
def smith_waterman_alignment(s1, s2, match_score=3, gap_cost=2):# 初始化得分矩阵A = np.zeros((len(s1) + 1, len(s2) + 1), int)for i, j in itertools.product(range(1, A.shape[0]), range(1, A.shape[1])):match = A[i - 1, j - 1] + (match_score if s1[i - 1] == s2[j - 1] else -match_score)delete = A[i - 1, j] - gap_costinsert = A[i, j - 1] - gap_costA[i, j] = max(match, delete, insert, 0)return np.max(A)# 编辑距离矩阵的计算
def compute_distance_matrix(reads):n_reads = len(reads)distance_matrix = np.zeros((n_reads, n_reads))for i in range(n_reads):for j in range(i+1, n_reads):alignment_score = smith_waterman_alignment(reads[i], reads[j])distance_matrix[i, j] = distance_matrix[j, i] = alignment_score # we use alignment score directly herereturn distance_matrix# Run SMITH-WATERMAN on the dataset
distance_matrix = compute_distance_matrix(reads['DNA'].values)# 聚类算法
def cluster_sequences(distance_matrix, n_clusters=2):# 使用层次聚类,可以使用其他聚类算法clustering = AgglomerativeClustering(n_clusters=n_clusters, affinity='precomputed', linkage='complete')# 使用 1 减 距离矩阵,是为了将距离转化为相似度clustering.fit(1 - distance_matrix)return clustering.labels_
# 聚类和评估
cluster_labels = cluster_sequences(distance_matrix)
reads['Cluster'] = cluster_labels# 评估簇的纯度
def evaluate_cluster_purity(cluster_labels, actual_labels):contingency_table = pd.crosstab(cluster_labels, actual_labels)purity = np.sum(np.max(contingency_table, axis=0)) / np.sum(contingency_table.sum())return purity# 可视化
def visualize_clustering(reads, cluster_labels):plt.figure(figsize=(12, 8))colors = ['r', 'g', 'b', 'y', 'c', 'm']for i in np.unique(cluster_labels):plt.plot(reads[reads['Cluster'] == i]['DNA'].index, [i] * sum(reads['Cluster'] == i), 'x', color=colors[i % len(colors)], label=f'Cluster {i}')plt.title('Clustering of DNA sequences')plt.xlabel('Sequence Index')plt.ylabel('Cluster ID')plt.legend()plt.show()visualize_clustering(reads, cluster_labels)方法三:对测序序列进行k-mer编码。使用CountVectorizer把序列的k-mer列表转换成词频(term frequency)矩阵。使用K-means算法对k-mer词频矩阵进行聚类,聚类数设置为原始序列数。
import pandas as pd
import numpy as np
from sklearn.cluster import KMeans
from sklearn.decomposition import PCA
from sklearn.feature_extraction.text import CountVectorizer
import matplotlib.pyplot as plt
import time# 读取数据
reference_seq_s = pd.read_csv('data/train_reference.txt', sep=' ', names=['ID', 'DNA_ref'])
reads = pd.read_csv('data/train_reads.txt', sep=' ', names=['ID', 'DNA'])# k-mer计数函数
def get_kmers(sequence, k=3):return [sequence[x:x+k] for x in range(len(sequence) + 1 - k)]reads['kmers'] = reads['DNA'].apply(lambda x: get_kmers(x))# 将k-mer列表转换为字符串(以便进一步转换为向量)
reads['kmers_str'] = reads['kmers'].apply(lambda x: ' '.join(x))# 使用CountVectorizer生成k-mer的词频矩阵
vectorizer = CountVectorizer()
X = vectorizer.fit_transform(reads['kmers_str'])# PCA降维
pca = PCA(n_components=2)
X_reduced = pca.fit_transform(X.toarray())# KMeans聚类
# 确定簇的数量为原始序列数
n_clusters = len(reference_seq_s['ID'].unique())
kmeans = KMeans(n_clusters=n_clusters)start_time = time.time()# 训练模型
kmeans.fit(X)
end_time = time.time()# 计算总耗时
total_time = (end_time - start_time) / 60
print("聚类时间{:.2f} minutes.".format(total_time))labels = kmeans.labels_
reads['cluster'] = labels
# 可视化结果
plt.figure(figsize=(8, 6))
plt.scatter(X_reduced[:, 0], X_reduced[:, 1], c=labels, cmap='rainbow', alpha=0.6, edgecolors='w', s=50)
plt.savefig('img/k-cluster.png',dpi=100)
plt.show()
3.3 问题三
import pandas as pd
from sklearn.cluster import KMeans
from sklearn.feature_extraction.text import CountVectorizer
import pyecharts.options as opts
from pyecharts.charts import Bar
import time# k-mer计数函数
def get_kmers(sequence, k=3):return [sequence[x:x+k] for x in range(len(sequence) + 1 - k)]
# 读取数据
# reference_seq_s = pd.read_csv('data/train_reference.txt', sep=' ', names=['ID', 'DNA'])
reads = pd.read_csv('data/train_reads.txt', sep=' ', names=['ID', 'DNA'])
test_reads = pd.read_csv('data/test_reads.txt',names=['DNA'])reads['kmers'] = reads['DNA'].apply(lambda x: get_kmers(x))
# 将k-mer列表转换为字符串(以便进一步转换为向量)
reads['kmers_str'] = reads['kmers'].apply(lambda x: ' '.join(x))
# 应用k-mer处理
test_reads['kmers'] = test_reads['DNA'].apply(lambda x: get_kmers(x))
test_reads['kmers_str'] = test_reads['kmers'].apply(lambda x: ' '.join(x))# 使用CountVectorizer生成k-mer的词频矩阵
vectorizer = CountVectorizer()
# 先拟合训练数据
X_train = vectorizer.fit_transform(reads['kmers_str'])
# 再转换测试数据
X_test = vectorizer.transform(test_reads['kmers_str'])from sklearn.decomposition import PCA
# 用PCA降维以便可视化(仅用于降维和可视化,并不用于聚类)
pca = PCA(n_components=2)
X_reduced = pca.fit_transform(X_train.toarray())
# KMeans聚类
start_time = time.time()
n_clusters = len(reads['ID'].unique())
kmeans = KMeans(n_clusters=n_clusters)
# 训练模型
kmeans.fit(X_train)
clusters = kmeans.fit_predict(X_test)
end_time = time.time()
# 输出聚类耗时
print(f"Clustering Time: {end_time - start_time}")# 统计每个簇的拷贝数
cluster_counts = pd.Series(clusters).value_counts().sort_index()


3.4 问题四
(1)方法一
from sklearn.decomposition import PCA
import pandas as pd
from sklearn.cluster import KMeans
from sklearn.feature_extraction.text import CountVectorizer
import time# k-mer计数函数
def get_kmers(sequence, k=3):return [sequence[x:x+k] for x in range(len(sequence) + 1 - k)]
# 读取数据
# reference_seq_s = pd.read_csv('data/train_reference.txt', sep=' ', names=['ID', 'DNA'])
reads = pd.read_csv('data/train_reads.txt', sep=' ', names=['ID', 'DNA'])
test_reads = pd.read_csv('data/test_reads.txt',names=['DNA'])reads['kmers'] = reads['DNA'].apply(lambda x: get_kmers(x))
# 将k-mer列表转换为字符串(以便进一步转换为向量)
reads['kmers_str'] = reads['kmers'].apply(lambda x: ' '.join(x))
# 应用k-mer处理
test_reads['kmers'] = test_reads['DNA'].apply(lambda x: get_kmers(x))
test_reads['kmers_str'] = test_reads['kmers'].apply(lambda x: ' '.join(x))
# 使用CountVectorizer生成k-mer的词频矩阵
vectorizer = CountVectorizer()
# 先拟合训练数据
X_train = vectorizer.fit_transform(reads['kmers_str'])
# 再转换测试数据
X_test = vectorizer.transform(test_reads['kmers_str'])
# 用PCA降维以便可视化(仅用于降维和可视化,并不用于聚类)
pca = PCA(n_components=2)
X_reduced = pca.fit_transform(X_train.toarray())
# KMeans聚类
start_time = time.time()
n_clusters = len(reads['ID'].unique())
kmeans = KMeans(n_clusters=n_clusters)
# 训练模型
kmeans.fit(X_train)
clusters = kmeans.fit_predict(X_test)# 比对模型的Python代码实现
import numpy as np
from collections import Counter
from typing import List# 函数来计算多数投票后确定的序列
def consensus_sequence(seqs: List[str]) -> str:"""采取多数投票法,返回一个列表中最可能正确的目标序列。:param seqs: 需要进行多数投票的一系列序列:return: 最可能正确的目标序列"""# 将序列转置,以方便进行列方向投票transposed_seqs = list(zip(*seqs))consensus_seq = []# 对于每个位置,计算最常见的元素for column in transposed_seqs:counter = Counter(column)most_common = counter.most_common(1)[0][0]consensus_seq.append(most_common)return ''.join(consensus_seq)# 根据聚类结果对序列进行聚类
clustered_seqs = {} # 存储每个原始序列ID对应的所有序列
# 对测试数据聚类
for idx, cluster_id in enumerate(clusters):if cluster_id not in clustered_seqs:clustered_seqs[cluster_id] = []clustered_seqs[cluster_id].append(test_reads['DNA'][idx])# 对于每个聚类,进行比对,并确定共识序列
consensus_seqs = {}
for cluster_id, seqs in clustered_seqs.items():consensus = consensus_sequence(seqs)consensus_seqs[cluster_id] = consensus
# 评估聚类质量和恢复的序列质量
reference_seqs = pd.read_csv('data/train_reference.txt', sep=' ', names=['ID', 'DNA'])# 评估聚类质量和恢复的序列质量
reference_seqs = pd.read_csv('data/train_reference.txt', sep=' ', names=['ID', 'DNA'])
# 计算共识序列与目标序列的错误率
def calculate_error_rate(original_seq: str, new_seq: str) -> float:"""计算恢复的序列与目标序列之间的错误率。:param original_seq: 原序列:param new_seq: 恢复的序列:return: 错误率"""errors = sum(1 for orig, new in zip(original_seq, new_seq) if orig != new)return errors / len(original_seq)# 错误率列表
error_rates = []
# 输出最可能正确的序列并计算错误率
for cluster_id, cons_seq in sorted(consensus_seqs.items()):original_seq = reference_seqs.loc[cluster_id,'DNA']error_rate = calculate_error_rate(original_seq, cons_seq)error_rates.append(error_rate)print(f"Cluster {cluster_id} Consensus: {cons_seq}, Error Rate: {error_rate}")# 分析总体错误率
overall_error_rate = np.mean(error_rates)
print(f"总体错误率: {overall_error_rate}")
总体错误率:0.509
(2)方法二
from sklearn.decomposition import PCA
import pandas as pd
from sklearn.cluster import KMeans
from sklearn.feature_extraction.text import CountVectorizer
import time# k-mer计数函数
def get_kmers(sequence, k=3):return [sequence[x:x+k] for x in range(len(sequence) + 1 - k)]
# 读取数据
# reference_seq_s = pd.read_csv('data/train_reference.txt', sep=' ', names=['ID', 'DNA'])
reads = pd.read_csv('data/train_reads.txt', sep=' ', names=['ID', 'DNA'])
test_reads = pd.read_csv('data/test_reads.txt',names=['DNA'])reads['kmers'] = reads['DNA'].apply(lambda x: get_kmers(x))
# 将k-mer列表转换为字符串(以便进一步转换为向量)
reads['kmers_str'] = reads['kmers'].apply(lambda x: ' '.join(x))
# 应用k-mer处理
test_reads['kmers'] = test_reads['DNA'].apply(lambda x: get_kmers(x))
test_reads['kmers_str'] = test_reads['kmers'].apply(lambda x: ' '.join(x))
# 使用CountVectorizer生成k-mer的词频矩阵
vectorizer = CountVectorizer()
# 先拟合训练数据
X_train = vectorizer.fit_transform(reads['kmers_str'])
# 再转换测试数据
X_test = vectorizer.transform(test_reads['kmers_str'])
# 用PCA降维以便可视化(仅用于降维和可视化,并不用于聚类)
pca = PCA(n_components=2)
X_reduced = pca.fit_transform(X_train.toarray())
# KMeans聚类
start_time = time.time()
n_clusters = len(reads['ID'].unique())
kmeans = KMeans(n_clusters=n_clusters)
# 训练模型
kmeans.fit(X_train)
clusters = kmeans.fit_predict(X_test)import numpy as np
import pandas as pd
from collections import Counter# Needleman-Wunsch算法实现
def needleman_wunsch(seq1, seq2, match_score=1, gap_cost=1, mismatch_cost=1):n = len(seq1)m = len(seq2)score_matrix = np.zeros((n+1, m+1))# Initialize score matrix and traceback pathsfor i in range(n+1):score_matrix[i][0] = -i * gap_costfor j in range(m+1):score_matrix[0][j] = -j * gap_cost# Fill in score matrixfor i in range(1, n+1):for j in range(1, m+1):if seq1[i-1] == seq2[j-1]:match = score_matrix[i-1][j-1] + match_scoreelse:match = score_matrix[i-1][j-1] - mismatch_costdelete = score_matrix[i-1][j] - gap_costinsert = score_matrix[i][j-1] - gap_costscore_matrix[i][j] = max(match, delete, insert)# Traceback to compute the alignmentalign1 = ""align2 = ""i = nj = mwhile i > 0 and j > 0:score_current = score_matrix[i][j]score_diagonal = score_matrix[i-1][j-1]score_up = score_matrix[i][j-1]score_left = score_matrix[i-1][j]if score_current == score_diagonal + (match_score if seq1[i-1] == seq2[j-1] else -mismatch_cost):align1 += seq1[i-1]align2 += seq2[j-1]i -= 1j -= 1elif score_current == score_left - gap_cost:align1 += seq1[i-1]align2 += "-"i -= 1elif score_current == score_up - gap_cost:align1 += "-"align2 += seq2[j-1]j -= 1while i > 0:align1 += seq1[i-1]align2 += "-"i -= 1while j > 0:align1 += "-"align2 += seq2[j-1]j -= 1return align1[::-1], align2[::-1]# 从聚类结果中恢复出最可能的序列
def recover_sequence(cluster_seqs):# 序列长度可能不同,先找到最长的序列长度略return consensus_sequencefrom functools import reduce
# 使用先前完成的KMeans结果clusters
# 假设clusters为序列的聚类结果,test_reads为相应的序列数据
cluster_dict = {i: [] for i in range(n_clusters)}
for i, cluster in enumerate(clusters):cluster_dict[cluster].append(test_reads['DNA'][i])# 对每个簇进行比对,并且输出最可能正确的序列
consensus_sequences = {}
for cluster_id, seqs in cluster_dict.items():if len(seqs) > 1:# 使用reduce函数将同簇序列两两比对consensus = reduce(lambda x, y: recover_sequence([x, y]), seqs)else:# 如果簇内只有一个序列,则将其作为最可能的序列consensus = seqs[0]consensus_sequences[cluster_id] = consensus# 将得到的“最可能正确的序列”写入到文件
with open('data/test_ref.txt', 'w') as f_out:for seq in consensus_sequences.values():f_out.write(seq + '\n')
4 完整代码
请看名片扣我
相关文章:
【2023年中国高校大数据挑战赛 】赛题 B DNA 存储中的序列聚类与比对 Python实现
【2023年中国高校大数据挑战赛 】赛题 B DNA 存储中的序列聚类与比对 Python实现 更新时间:2023-12-29 1 题目 赛题 B DNA 存储中的序列聚类与比对 近年来,随着新互联网设备的大量涌入和对其服务需求的指数级增长,越来越多的数据信息被产…...
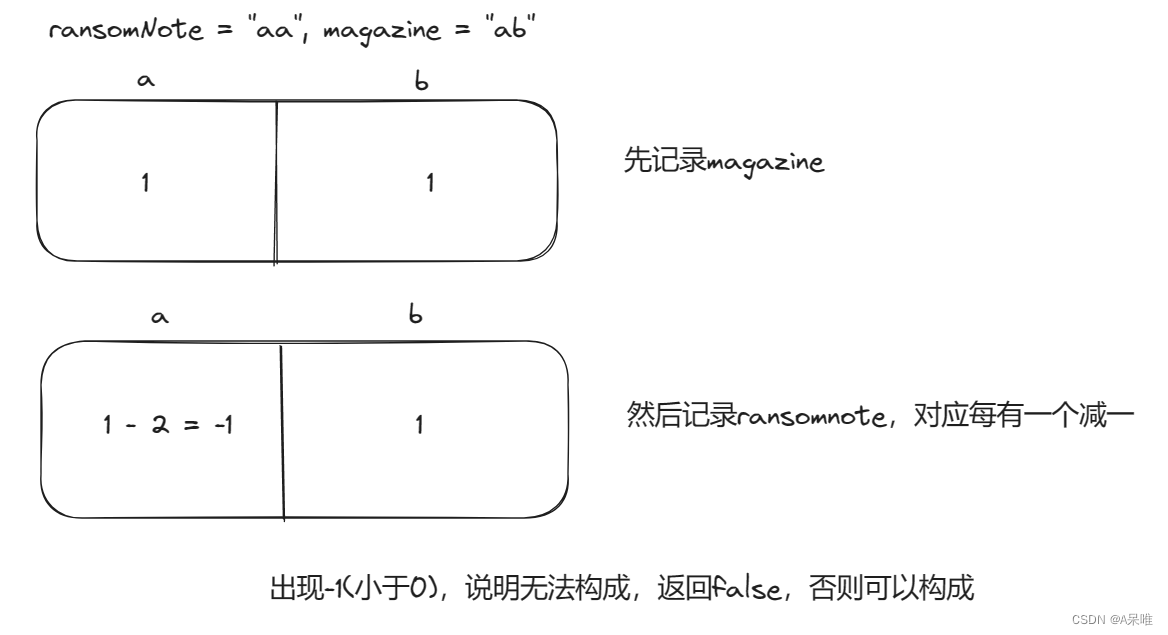
力扣383.赎金信 -- 哈希表
思路:记录magazine每个字符个数,然后记录ransomNote每个字符(每有一个减1),假如出现<0的情况说明ransomnode有字符的个数超过了magazine则无法构成,否则可以构成 代码: class Solution { pu…...
GeoServer发布地图服务(WMS、WFS)
文章目录 1. 概述2. 矢量数据源3. 栅格数据源 1. 概述 我们知道将GIS数据大致分成矢量数据和栅格数据(地形和三维模型都是兼具矢量和栅格数据的特性)。但是如果用来Web环境中,那么使用图片这个栅格形式的数据载体无疑是最为方便的࿰…...

C语言——结构体
一、结构体的创建 1、定义 在 C 语言中,结构体是一种自定义的数据类型,它允许将不同类型的数据项组合成一个单一实体。这在组织复杂数据时非常有用,因为它可以将有逻辑关系的数据组合在一起。结构体是一些值的集合,这些值是结构…...
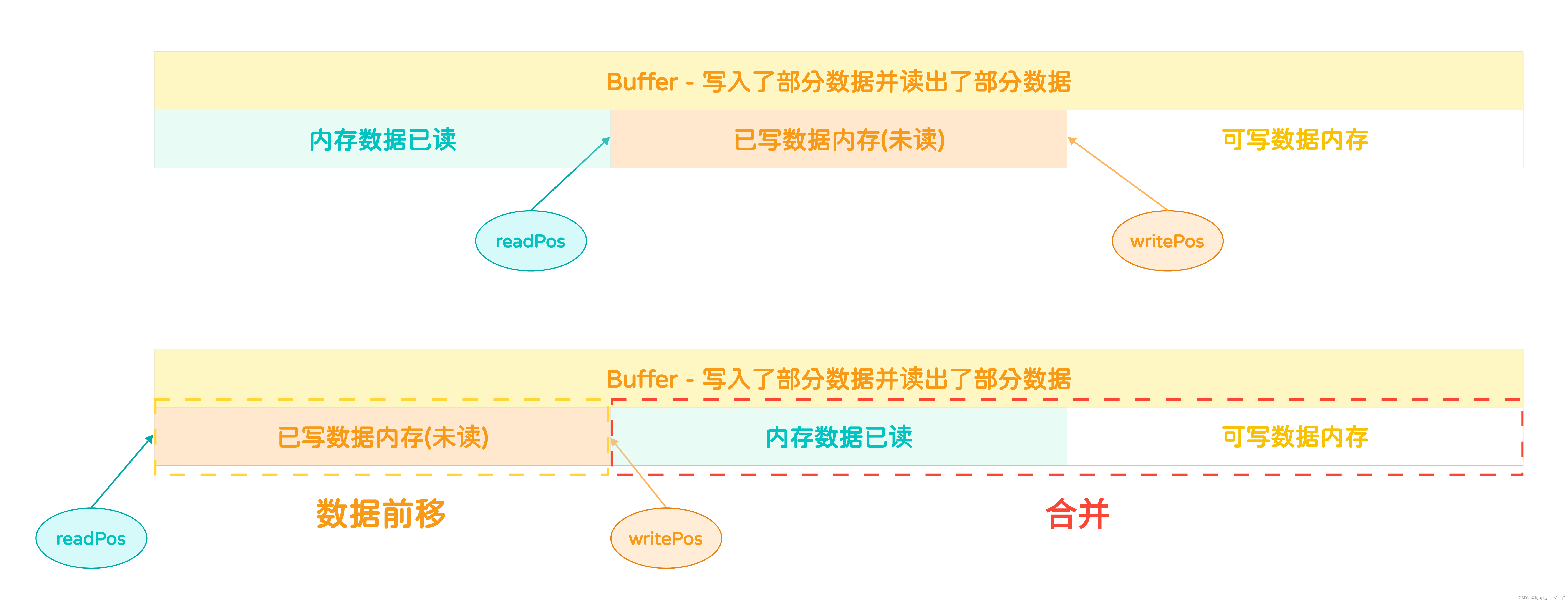
基于多反应堆的高并发服务器【C/C++/Reactor】(中)Buffer的创建和销毁、扩容、写入数据
TcpConnection:封装的就是建立连接之后得到的用于通信的文件描述符,然后基于这个文件描述符,在发送数据的时候,需要把数据先写入到一块内存里边,然后再把这块内存里边的数据发送给客户端,除了发送数据,剩下…...

【Linux】常用的基本命令指令①
前言:从今天开始,我们逐步的学习Linux中的内容,和一些网络的基本概念,各位一起努力呐! 💖 博主CSDN主页:卫卫卫的个人主页 💞 👉 专栏分类:数据结构 👈 💯代码…...

活动运营常用的ChatGPT通用提示词模板
活动目标确定:如何明确活动的目标,确保活动策划与执行的方向性? 活动主题选择:如何选择吸引人的活动主题,提高用户的参与度和兴趣? 活动形式策划:如何根据活动目标和主题,选择适合…...

SpringBoot 中实现订单30分钟自动取消的策略
简介 在电商和其他涉及到在线支付的应用中,通常需要实现一个功能:如果用户在生成订单后的一定时间内未完成支付,系统将自动取消该订单。 本文将详细介绍基于Spring Boot框架实现订单30分钟内未支付自动取消的几种方案,并提供实例…...

像专家一样使用TypeScript映射类型
掌握TypeScript的映射类型,了解TypeScript内置的实用类型是如何工作的。 您是否使用过Partial、Required、Readonly和Pick实用程序类型? 你知道他们内部是怎么运作的吗? 如果您想彻底掌握它们并创建自己的实用程序类型,那么不要错过本文所涵盖的内容。…...

Golang 结构体
前言 在 Go 语言中,结构体(struct)是一种自定义的数据类型,将多个不同类型的字段(fields)组合在一起 结构体通常用于模拟真实世界对象的属性和行为 定义结构体 可以使用 type 关键字和 struct 关键字来定…...

服务器运行状况监控工具
服务器运行状况监视提供了每个服务器状态和性能的广泛概述,通过监控服务器指标,如 CPU 使用率、内存消耗、I/O、磁盘使用率、进程等,服务器运行状况监控可以避免服务器停机。 服务器性能监控指标 服务器是网络中最重要的组件之一࿰…...
)
2022年全国职业院校技能大赛软件测试赛题卷②—自动化测试解析报告(含术语)
2022年全国职业院校技能大赛软件测试任务四 自动化测试 目录 第一题:按照以下步骤在PyCharm中进行自动化测试脚本编写,并执行脚本。...

497 蓝桥杯 成绩分析 简单
497 蓝桥杯 成绩分析 简单 //C风格解法1,*max_element()与*min_element()求最值 //时间复杂度O(n),通过率100% #include <bits/stdc.h> using namespace std;using ll long long; const int N 1e4 …...
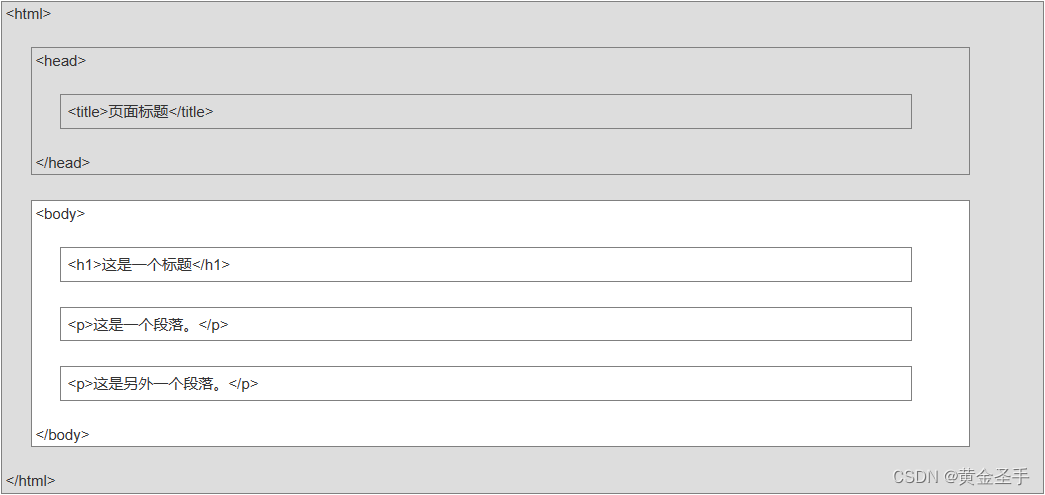
一、HTML5简介
一、简介 超文本标记语言(英语:HyperText Markup Language,简称:HTML)是一种用于创建网页的标准标记语言。可以使用 HTML 来建立自己的 WEB 站点,HTML 运行在浏览器上,由浏览器来解析。 <!…...
视频云存储/视频智能分析平台EasyCVR在麒麟系统中无法启动该如何解决?
安防视频监控/视频集中存储/云存储/磁盘阵列EasyCVR平台可拓展性强、视频能力灵活、部署轻快,可支持的主流标准协议有国标GB28181、RTSP/Onvif、RTMP等,以及支持厂家私有协议与SDK接入,包括海康Ehome、海大宇等设备的SDK等。平台既具备传统安…...
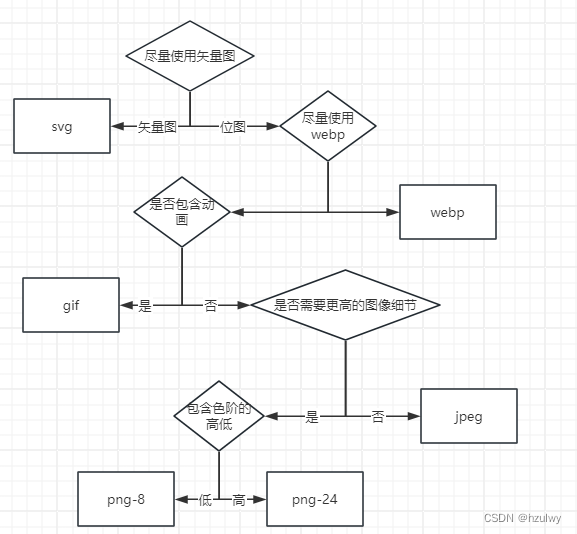
前端性能优化之图像优化
图像优化问题主要可以分为两方面:图像的选取和使用,图像的加载和显示。 图像基础 HTTP Archive上的数据显示,网站传输的数据中,60%的资源都是由各种图像文件组成的,当然这些是将各类型网站平均的结果,单独…...
微信小程序封装vant 下拉框select 单选组件
先上效果图: 主要是用vant 小程序组件封装的:vant 小程序ui网址:vant-weapp 主要代码如下: 先封装子组件: select-popup 放在 components 文件夹里面 select-popup.wxml: <!--pages/select-popup/select-popup.wxml--> &…...

c语言试卷
江西财经大学IT帮 2020-2021第一学期期末C语言模拟考试试卷 课程名称:C语言程序设计(软件)(主干课程) 适用对象:21级本科 试卷命题人 钟芳盛 游天悦 李俊贤 万军豪 张位 试卷审核人 钟芳盛 一、单项…...

文献阅读:Sparse Low-rank Adaptation of Pre-trained Language Models
文献阅读:Sparse Low-rank Adaptation of Pre-trained Language Models 1. 文章简介2. 具体方法介绍 1. SoRA具体结构2. 阈值选取考察 3. 实验 & 结论 1. 基础实验 1. 实验设置2. 结果分析 2. 细节讨论 1. 稀疏度分析2. rank分析3. 参数位置分析4. 效率考察 4.…...
NCC基础开发技能培训
YonBuilder for NCC 是一个带插件的eclipse工具,跟eclipse没什么区别 NC Cloud2021.11版本开发环境搭建改动 https://nccdev.yonyou.com/article/detail/495 不管是NC Cloud 新手还是老NC开发,在开发NC Cloud时开发环境搭建必看!ÿ…...

开源工具Cursor Free VIP:突破AI编程限制的高效使用指南
开源工具Cursor Free VIP:突破AI编程限制的高效使用指南 【免费下载链接】cursor-free-vip [Support 0.45](Multi Language 多语言)自动注册 Cursor Ai ,自动重置机器ID , 免费升级使用Pro 功能: Youve reached your t…...

滑动窗口-438. 找到字符串中所有字母异位词
文章目录1.题解核心解题思路(滑动窗口)2.机考代码3.知识点讲解1. map.getOrDefault(key, defaultValue)2. map.put(key, value)3. map.containsKey(key)4. s.toCharArray()5. s.charAt(index)6. Scanner 相关(机考必备)力扣地址&a…...

QMCFLAC2MP3深度解析:从格式解密到跨设备音频转换的全流程实践
QMCFLAC2MP3深度解析:从格式解密到跨设备音频转换的全流程实践 【免费下载链接】qmcflac2mp3 直接将qmcflac文件转换成mp3文件,突破QQ音乐的格式限制 项目地址: https://gitcode.com/gh_mirrors/qm/qmcflac2mp3 问题引入:破解音乐格式…...

如何永久保存微信聊天记录:WeChatMsg本地化解决方案
如何永久保存微信聊天记录:WeChatMsg本地化解决方案 【免费下载链接】WeChatMsg 提取微信聊天记录,将其导出成HTML、Word、CSV文档永久保存,对聊天记录进行分析生成年度聊天报告 项目地址: https://gitcode.com/GitHub_Trending/we/WeChatM…...

文墨共鸣大模型与Matlab科学计算结合:数据报告自动化
文墨共鸣大模型与Matlab科学计算结合:数据报告自动化 每次做完仿真和数据分析,看着满屏的图表和密密麻麻的数据矩阵,你是不是也头疼怎么写报告?从数据到文字,这中间仿佛隔着一道鸿沟,既要组织语言…...

突破性数字音乐解放方案:QMCDecode实战指南与3大智能转换场景解密
突破性数字音乐解放方案:QMCDecode实战指南与3大智能转换场景解密 【免费下载链接】QMCDecode QQ音乐QMC格式转换为普通格式(qmcflac转flac,qmc0,qmc3转mp3, mflac,mflac0等转flac),仅支持macOS,可自动识别到QQ音乐下载目录&#…...

实测联想小新Pro 16 GT:一台把性能、AI和续航拉满的AI PC
最近体验了联想小新Pro 16 GT AI元启版,它不像是传统轻薄本,更像一台兼顾便携、性能和智能体验的全能机型。抛开品牌滤镜,单看硬件和实际使用,确实有不少值得一说的亮点。外观轻薄耐看,屏幕和接口都很实在这台机器用了…...

ncmdumpGUI:网易云音乐NCM文件转换完全解决方案
ncmdumpGUI:网易云音乐NCM文件转换完全解决方案 【免费下载链接】ncmdumpGUI C#版本网易云音乐ncm文件格式转换,Windows图形界面版本 项目地址: https://gitcode.com/gh_mirrors/nc/ncmdumpGUI 为什么你的付费音乐无法自由播放?——N…...

【架构心法】撕碎“实验室完美”的傲慢!直视滚刀与高压现场的物理混沌,论工业级控制系统的“防御性悲观主义”
摘要:在纯净的实验室里,“1”永远是“1”,“0”永远是“0”。但在重型机械的施工现场,物理法则充满了不可预测的恶意。无数工程师带着“代码没 Bug 就不会死机”的天真走向现场,最终却在震动、高温与电磁噪声的围剿下全…...

Graphormer开源模型多场景落地:高校科研、药企CADD、新材料研发实操路径
Graphormer开源模型多场景落地:高校科研、药企CADD、新材料研发实操路径 1. 项目概述 Graphormer是一种基于纯Transformer架构的图神经网络模型,专门为分子图(原子-键结构)的全局结构建模与属性预测而设计。该模型在OGB、PCQM4M…...